A 61-year-old Indian man was admitted to the hospital due to weakness of his left upper and lower extremities and throbbing, frontal headache which began a day prior admission. The patient experienced fatigue and difficulty in standing and walking. These were not associated with fever, nausea, vomiting, dizziness, seizure, syncope, palpitations or chest discomfort. He also complained of intermittent lower limb cramp over the last 6 months.
The patient was diagnosed to have bilateral eye cataract and scheduled for corrective surgery after complaining of blurring of vision for the past 6 months. There was no history of giddiness, palpitation, chest discomfort or fainting episode. He also did not have any fever or any constitutional symptom. His appetite was normal and there was no change in his body weight. He denied any history of reduced libido nor erectile dysfunction. He had no history of polydipsia or polyuria. He has no tremors, heat and cold intolerance, and changes in bowel habit.
There was no history of previous head trauma or irradiation. He had no perinatal complications and developmental delay. He was diagnosed to have essential hypertension 10 years ago and is on amlodipine 5 mg once a day. He did not take any traditional medication. He had cleft palate and lip corrective surgery done at the age of 17 years old which left him with residual nasal speech.
The patient was a non-smoker and did not take any illicit drug or consumed alcohol. His family history was unremarkable for any endocrine disorder. He was married with 7 children. He was unemployed.
On examination, the patient was alert and oriented with coherent nasal speech and overweight. His vital signs were stable with blood pressure of 140/70 mm Hg with no evidence of postural hypotension, pulse rate 80 beats/minute, respiratory rate 18 breaths/min and afebrile. There was no goiter in neck examination. He did not have any features to suggest Cushing’s syndrome or acromegaly. His axillary and public hairs were not sparse and testicular sizes were normal. There was neither oral candidiasis nor any skeletal deformities. There was no evidence of involuntary movements or bilateral carpo-pedal spasm. Chvostek’s and Trousseau’s signs were negative. His vision was 6/12 in both eyes with presence of bilateral cataract. Visual field confrontation test revealed bitemporal hemianopia. Cranial nerve examination was normal. Muscle tone was normal, and there were no fasciculation or atrophy in any muscle group. Strength was normal in distal muscle and slightly reduced in the left-sided pelvic and scapular girdles. Deep tendon reflexes were normal. Cardiovascular examinations were essentially normal with no cardiac murmur heard during auscultation. The rest of the physical examination was unremarkable.
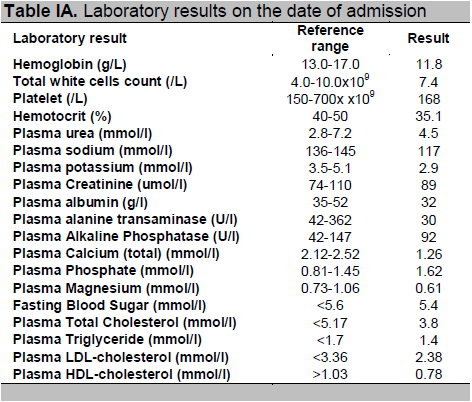
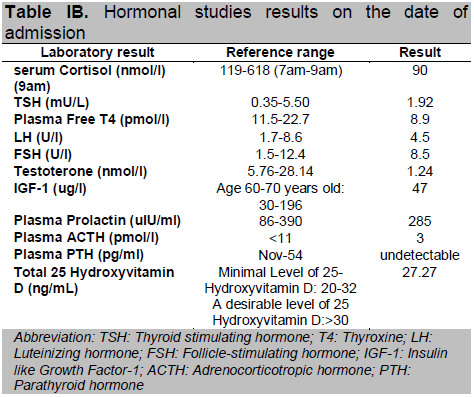
Routine hematological tests and liver function tests were normal (Table 1A). However, the biochemical tests showed multiple electrolyte abnormalities (hyponatremia, hypokalemia, hypomagnesemia, severe hypocalcemia and hyperphosphatemia) and normal blood levels of albumin, creatinine and alkaline phosphatase (Table 1A). Magnesium level was slightly decreased. The serum level of intact parathyroid hormone (iPTH) was undetectable and the 25-hydroxyvitamin D level was borderline low (Table 1B). Low free thyroxine (FT4) and inappropriately normal thyroid stimulating hormone (TSH) were suggestive of secondary hypothyroidism. Investigations for causes of hyponatraemia revealed that this patient had hypocortisolism and inappropriate response of thyrotrophin stimulating hormone (TSH) suggestive of secondary hypothyroidism. Further hormonal tests are suggestive of secondary hypogonadism with testosterone level of 1.24 nmol/l, follicle stimulating hormone (FSH) 8.5 U/I, luteinizing hormone (LH) 4.5 U/l, and lower normal range of insulin growth factor (IGF-1) (Table 1B). Electrocardiography was normal, with no evidence of prolonged QT interval. Transthoracic echocardiography revealed normal ejection fraction (61%) and chamber sizes, with no evidence of left ventricular hypertrophy or any abnormalities suggestive of congenital heart disease and valvular abnormalities noted.
Click here to download Table IA
Table IA. Laboratory results on the date of admission
Click here to download Table IB
Table IB. Hormonal studies results on the date of admission
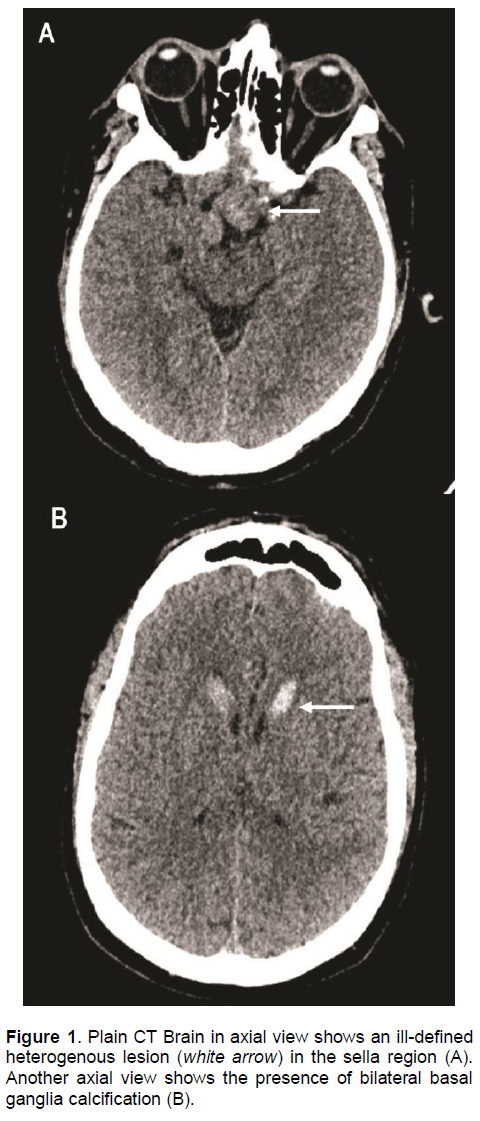
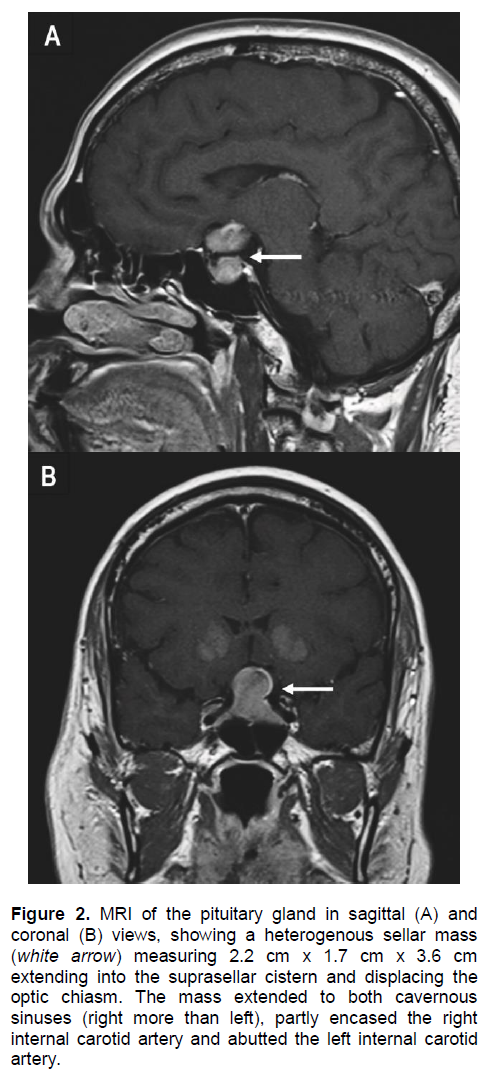
An urgent computed tomography (CT) scan of the brain revealed a suspicious sellar lesion together with bilateral head of caudate nucleus and basal ganglia calcification (Figures 1A and 1B). Magnetic resonance imaging (MRI) of the pituitary demonstrated a heterogeneous sellar mass measuring 2.2 x 1.7 x 3.6 cm extending into the suprasellar cistern displacing the optic chiasm. There was also extension to both cavernous sinuses (right more than left), partly encasing the right internal carotid artery and abuts the left internal carotid artery (Figures 2A and 2B).
Click here to download Figure 1
Figure 1 . Plain CT Brain in axial view shows an ill-defined heterogenous lesion (white arrow) in the sella region (A). Another axial view shows the presence of bilateral basal ganglia calcification (B).
Click here to download Figure 2
Figure 2. MRI of the pituitary gland in sagittal (A) and coronal (B) views, showing a heterogenous sellar mass (white arrow) measuring 2.2 cm x 1.7 cm x 3.6 cm extending into the suprasellar cistern and displacing the optic chiasm. The mass extended to both cavernous sinuses (right more than left), partly encased the right internal carotid artery and abutted the left internal carotid artery.
A diagnosis of non-functioning pituitary macroadenoma with panhypopituitarism and primary hypopara-thyroidism was made based on the biochemical tests and imaging studies. The patient was given levothyroxine, hydrocortisone, calcitriol and calcium tablet. He eventually underwent transsphenoidal hypophysectomy for his pituitary adenoma. His postoperative course was uneventful. Histopathologic examination of the sellar tumor did not reveal mitotic figures, lymphoplasmacytic infiltration or expression of growth hormone, prolactin, adrenocorticotropic hormone (ACTH), TSH, LH and FSH. No evidence of lymphoplasmacytic infiltration suggest lymphocytic hypophysitis in the histopathology examination. He continues to require hormone replacement therapy including the thyroxine, steroid, testosterone and active vitamin D.
A possibility of an autoimmune condition such as lymphocytic hypophysitis was suspected in view of the coexistence of primary hypoparathyroidism and pituitary tumor. However, the anti thyroglobulin antibody is 2.58IU/ml (negative if <120IU/ml), anti thyroid peroxidase is 1.04IU/ml (negative is ≤40IU/ml) are both within normal range. Pituitary antibodies were not sent as it was not available in our local laboratory. In view of his history of cleft lip and palate, together with primary hypo-parathyrodism there is also a possibility of velo-cardio-facial syndrome although his cardiovascular and echocardiography examinations were normal.
The clinical features, laboratory results, hormonal survey and imaging studies were consistent with primary hypoparathyroidism and hypopituitarism. The presentation of headache, visual impairments and defects, pituitary insufficiency together with the detection of a sellar mass is supportive of the diagnosis of a non-functioning pituitary macroadenoma. The etiology of primary hypoparathyroidism is most likely idiopathic primary hypoparathyroidism, in view of undetectable serum parathyroid hormone with concomitant hypocalcemia and hyperphosphatemia.
The coexistence of primary hypoparathyroidism and hypopituitarism secondary to a pituitary tumor in this patient may have been coincidental. The association of pituitary tumors and primary hyperparathyroidism has been established in multiple endocrine neoplasia type 1 (MEN1), but not for primary hypoparathyrodism. The association of autoimmune primary hypoparathyroidism with other endocrine and non-endocrine diseases has been described in polyglandular autoimmune syndrome (PAS). The association of hypoparathyroidism and hypo-pituitarism may occur in PAS type II.1 The prevalence of PAS type II is 1:20,000, where primary hypopara-thyroidism occurs in 3% and hypopituitarism between 0 to 2%.2 It is more frequently encountered in women, at a ratio of 1:3.3 There are only a few case reports on coexistent primary hypoparathyroidism and hypopituitarism. Arvanitakis and Knouss reported a case of selective hypopituitarism in association with primary hypoparathyroidism and mucocutaneous candidiasis.4 Salti and colleagues described a patient with primary hypoparathyroidism and hypopituitarism of unknown etiology due to absence of histological evidence.5
It is reasonable to postulate lymphocytic hypophysitis (LYH) as the differential diagnosis of the pituitary lesion associated with the autoimmune primary hypopara-thyrodism. It is a rare autoimmune endocrine disorder of pituitary6 affecting women more frequently than men, with a male-to-female ratio of 1:8.7 The clinical features are very similar to pituitary adenoma which is the mass related effect,8 cranial diabetes insipidus9 and hypopituitarism. Our patient had presented with similar symptoms and the hormonal studies are suggestive of hypopituitarism. In pituitary MRI, patients with LYH usually show a pituitary enlargement with symmetrical suprasellar extension which can displace the optic chiasms, whereas patients with adenoma show asymmetrical pituitary enlargement. The pituitary stalk may be deviated in the adenoma but is usually thickened in LYH.10 The pituitary enhancement after injection of gadolinium is homogeneously intense in LYH and shows a strip of enhanced tissue along the dura madre (also called ‘dural tail’). Patients with adenomas instead show delayed and poor enhancement, usually without a dural tail after gadolinium.9 The asymmetrical enlargement of pituitary tumor demonstrated in our MRI pituitary (Figure 2) for this patient is more consistent with pituitary macroadenoma.
LYH is frequently associated with other endocrine and non-endocrine autoimmune diseases9 such as Hashimoto’s thyroiditis or Graves’ disease. 12 Until now, anti-pituitary antibodies (APA) have not been considered good markers of LYH because of various difficulties in methodology and clinical interpretation.10 In view of the most common association which is Hashimoto’s thyroiditis or Graves’ disease, we tested for anti-thyroglobulin antibody and anti-thyroid peroxidase, but both results were within normal range. Currently, the diagnosis of LYH is still reliant on a high index of clinical suspicion as well as histopathological findings from pituitary biopsy which show lymphoplasmacytic infiltrate with lymphoid aggregates surrounding atrophic acini of pituitary cells.9 In our patient, the histopathologic analysis of tumor resected from transphenoidal hypophysectomy confirmed the diagnosis of non-functioning pituitary macroadenoma.
Velocardiofacial syndrome (VCFS) is an autosomal dominant disorder caused by a deletion in chromosome 22q11 which is also identified in majority of patients with DiGeorge syndrome.13 The description of the possible facial abnormalities in DGS include low set and posteriorly rotated ears, ocular hypertelorism, and palatal anomalies. Conotruncal cardiac defects are present in approximately 80% of DGS patients. Hypocalcemia, resulting from underdevelopment of the parathyroid glands, has been described in the newborn period in up to 60% of DGS patients. The complications of hypocalcemia are unusual later in life secondary to presumed compensatory hyperplasia of existing parathyroid tissue. However, hypocalcemia can be precipitated by extreme stress in older patients. Despite the patient’s history of cleft palate and concurrent hypocalcemia, he does not have the characteristic facial features of VCFS. He also had normal echocardiographic findings.
The case we described here has definite clinical, biochemical, radiological and histopathologic evidence to support the diagnosis of primary hypoparathyroidism and non-functioning pituitary macroadenoma.
Some of the presenting complaints of primary hypoparathyroidism and non-functioning pituitary adenoma are nonspecific and frequently not appreciated, therefore erroneously attributed to other causes. Thus, meticulous history taking; a thorough clinical examination and careful interpretation of laboratory results based on available evidence will often lead to the correct diagnosis. Although the association may be incidental, we feel that it is important to recognize the combination of endocrine tumors, as screening for other associated endocrine neoplasms is indicated.
1. Kahaly GJ. Polyglandular autoimmune syndromes. Eur J Endocrinol. 2009;161(1):11-20.
2. Ten S, New M, Maclaren N. Clinical review 130: Addison’s disease 2001. J Clin Endocrinol Metab. 2001;86(7):2909-22.
3. Fӧrster G, Krummenauer F, Kühn I et al. Polyglandular autoimmune syndrome type II: Epidemiology and forms of manifestation. Dtsch Med Wochenschr .1999;124(49):1476-81.
4. Arvanitakis C, Knouss RF. Selective hypopituitarism. Impaired cell-mediated immunity and chronic mucocutaneous candidiasis. JAMA. 1973;225(12):1492-5.
5. Salti IS, Mouradian A, Amiri Z et al. Hypopituitarism in a patient with idiopathic hypoparathyroidism. Can Med Assoc J. 1982;126(8):942-3.
6. Pestell RG, Best JD, Alford FP. Lymphocytic hypophysitis. The clinical spectrum of the disorder and evidence for an autoimmune pathogenesis. Clin Endocrinol (Oxf). 1990;33(4):457-66.
7. Durán Martinez M, Santonja C, Pavón de Paz I et al. Lymphocytic hypophysitis: Report of an unusual case of a rare disorder. J Endocrinol Invest. 2001;24(3):190-3.
8. Beressi N, Beressi JP, Cohen R et al. Lymphocytic hypophysitis. A review of 145 cases. Ann Med Interne (Paris). 1999;150(4):327-41.
9. Bellastella A, Bizzarro A, Coronella C et al. Lymphocytic hypophysitis: A rare or underestimated disease. Eur J Endocrinol. 2003;149(5):363-76.
10. Ahmadi J, Myers GS, Segall HD et al. Lymphocytic adenophypophysis: Contrast-enhanced MR imaging in five cases. Radiology. 1995;195(1):30-4.
11. Barbaro D, Loni G. Lymphocytic hypophysitis and autoimmune thyroid disease. J Endocrinol Invest. 2000;23(5):339-40.
12. De Bellis A, Bizzarro A, Bellastella A. Pituitary antibodies and lymphocytic hypophysitis. Best Pract Res Clin Endocrinol Metab. 2005;19(1);67-84.
13. McDonald-McGinn DM, Sullivan KE. Chromosome 22q11.2 deletion syndrome (DiGeorge syndrome/velocardiofacial syndrome). Medicine (Baltimore). 2011;90(1):1-18.
14. Hiéronimus S, Bec-Roche M, Pedeutour F et al. The spectrum of parathyroid gland dysfunction associated with the microdeletion 22q11. Eur J Endocrinol. 2006;155(1):47-52.
Articles and any other material published in the JAFES represent the work of the author(s) and should not be construed to reflect the opinions of the Editors or the Publisher.
Authors are required to accomplish, sign and submit scanned copies of the JAFES Declaration that the article represents original material that is not being considered for publication or has not been published or accepted for publication elsewhere.
Consent forms, as appropriate, have been secured for the publication of information about patients; otherwise, authors declared that all means have been exhausted for securing such consent.
The authors have signed disclosures that there are no financial or other relationships that might lead to a conflict of interest. All authors are required to submit Authorship Certifications that the manuscript has been read and approved by all authors, and that the requirements for authorship have been met by each author.