Infundibuloneurohypophysitis is a rare disorder which may present acutely with central diabetes insipidus. It may be familial, idiopathic or secondary to an autoimmune inflammatory process. In previous case reports, the condition was observed to occur in the immediate postpartum period.1 We report a case of infundibuloneurohypophysitis presenting later at 2 years postpartum.
A 31-year-old Indian woman presented with a 6-month history of polyuria and polydipsia. She had a reported urine volume of more than 6 liters per day. Two years before the onset of her symptoms, she had pregnancy-induced hypertension necessitating termination of the pregnancy by Caesarean section. Her menstruation restarted 6 months after delivery. She took cyclic drospirenone/ethinyl estradiol tablets which effectively regulated her menstrual cycle. She discontinued the medication 3 months prior to the onset of symptoms. She had no other medications. Review of systems and family history were unremarkable.
Physical examination was unremarkable. Visual fields were intact on confrontation testing. Baseline tests revealed a serum osmolality and serum sodium were both normal, in the presence of a very dilute and hypotonic urine (Table 1). Anterior pituitary hormonal profile revealed hyperprolactinemia [serum prolactin level 1626 mIU/L, polyethylene glycol (PEG) prolactin 79%] and gonadotropin levels consistent with the luteal phase of the menstrual cycle (Table 1). Corrected calcium, fasting plasma glucose, glycated hemoglobin, thyroid stimulating hormone, free thyroxine, random serum cortisol and adrenocorticotropic hormone levels were normal.
Click here to download Table 1
Table 1. Baseline laboratory results
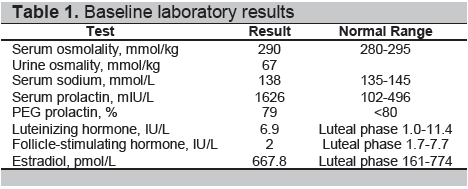
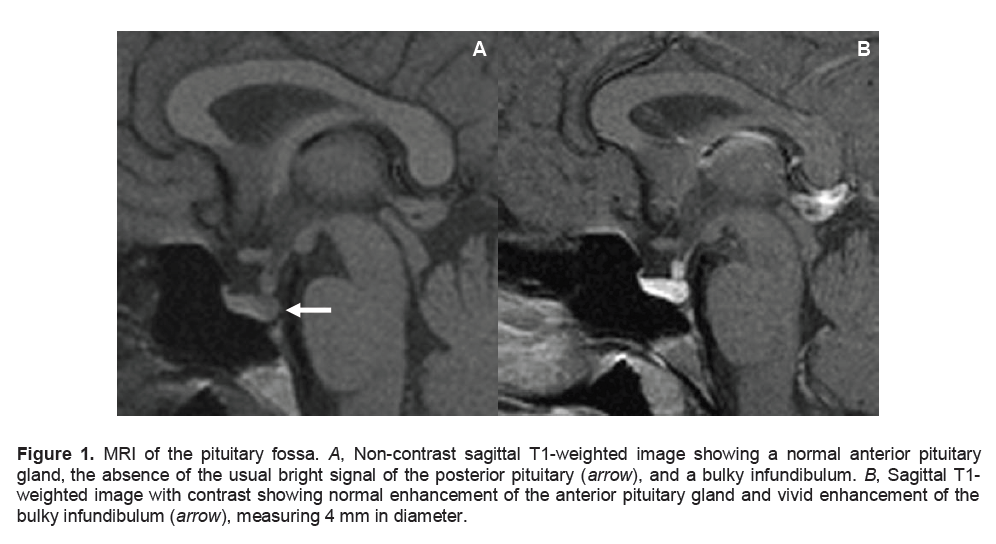
The results of the water deprivation test confirmed the diagnosis of central diabetes insipidus. At a serum osmolality of 303 mmol/kg, urine osmolality remained low at 71 mmol/kg. After desmopressin 2 μg was given subcutaneously, serum osmolality decreased to 286mmol/kg and urine osmolality increased to 611 mmol/kg (Table 2). Skull radiograph was unremarkable. A Magnetic Resonance Imaging (MRI) of the pituitary fossa revealed a normal anterior hypophysis, with a small (4 mm x 2 mm) nodular enhancing appearance of the lower part of the pituitary infundibulum associated with ectopia of the posterior pituitary gland, compatible with infundibuloneurohypophysitis (Figures 1 and 2).
Click here to download Figure 1
Figure 1. MRI of the pituitary fossa. A, Non-contrast sagittal T1-weighted image showing a normal anterior pituitary gland, the absence of the usual bright signal of the posterior pituitary (arrow), and a bulky infundibulum. B, Sagittal T1-weighted image with contrast showing normal enhancement of the anterior pituitary gland and vivid enhancement of the bulky infundibulum ( arrow), measuring 4 mm in diameter.
Click here to download Figure 2
Figure 2. Coronal MRI of the pituitary fossa. C, Non-contrast T1-weighted image showing a normal anterior pituitary and a bulky infundibulum (arrow). D, T1-weighted image with contrast showing normal enhancement of the anterior pituitary gland and vivid enhancement of the bulky infundibulum (arrow), measuring 4 mm in diameter.
Click here to download Table2
Table 2. Water deprivation test results

Further work-up to exclude other differential diagnoses was performed. A 25-hydroxy vitamin D assay to screen for sarcoidosis was low (6.6 μg/L). Cerebrospinal fluid examination revealed normal cytology and β-HCG level. Chest radiograph was unremarkable.
The patient was started on oral desmopressin 100 μg at bedtime and ergocalciferol 50,000 IU weekly. Menstruation became regular during her inpatient stay. During her last clinic visit, she reported improvement in symptoms. Oral desmopressin dose was maintained.
Lymphocytic hypophysitis (LYH) is an autoimmune condition characterized by lymphocytic infiltration of the pituitary gland accompanied by variable degrees of pituitary dysfunction. The clinical presentation is variable, depending on whether it affects the anterior hypophysis (lymphocytic adenohypophysitis, LAH) with early destruction of ACTH-producing cells; or the infundibulum, pituitary stalk and neurohypophysis (lymphocytic infundibuloneurohypophysitis, LINH); or an extensive anterior and posterior pituitary involvement (lymphocytic infundibulopanhypophysitis, LIPH).2 As with most autoimmune conditions, it occurs more frequently in women.1 LYH has been reported to be associated with other autoimmune conditions in 20 to 50% of cases. 3,4 Animal models have also shown association with serum autoantibodies against pituitary cells.5 In our patient, there were no signs of associated autoimmune disease.
Eighty to 90% of reported LAH cases are women. Of these, 90% were found to occur premenopause, with 50 to 75% presenting during the peripartum period. 1,5,6 The mean age at diagnosis is 34.5 years for females and 44.7 years for males. On the other hand, LINH exhibits a balanced sex distribution. The mean age at presentation is 47.3 years, older than the observed age in patients with LAH.2 Our patient presented with symptoms 2 years postpartum when she was 31 years old.
LINH is the most likely diagnosis in cases of suspected LYH, with diabetes insipidus as the presenting or most prominent condition. There may also be mass-effect symptoms with headaches or other pituitary hormone deficiencies, although anterior pituitary function is frequently intact. Adenohypophyseal involvement is usually mild and transient, most commonly involving growth hormone, and less commonly gonadotropins and thyrotropin. Prolactin level is usually normal or mildly elevated. Sudden onset of diabetes insipidus is also seen in 14 to 20% of LAH cases, attributed to inflammation, destruction of the posterior pituitary or pituitary stalk, or surgery.2 Our patient had diabetes insipidus and hyperprolactinemia, with sparing of other anterior pituitary functions.
LYH is thought to progress from inflammation to fibrosis and subsequent atrophy, eventually manifesting as an empty sella. However, spontaneous resolution has been documented.1 The initial inflammation with gland enlargement may present as a mass and may lead to permanent hypopituitarism. Some cases are associated with an insidious or a relapsing-remitting course. In LINH, inflammation is self-limited, with radiological resolution after about 2 years.1 However, complete or partial central diabetes insipidus can be permanent secondary to neuronal destruction.
The differential diagnoses of diabetes inspidus may be further investigated by MRI of the pituitary and the hypothalamus. The normal functioning posterior pituitary presents as a hyperintense signal or bright spot. The source of the hyperintense signal seen in the posterior pituitary gland on T1-weighted images remains controversial. However, it is generally accepted as a functional marker of the neurohypophysis and is noted to be absent in central diabetes inspidus. The bright spot can be detected in nearly 100% of healthy persons when high-resolution MRI is used.7 Most routine brain imaging protocols, on the other hand, do not provide high-resolution sequences of the sella. As such, the posterior pituitary bright spot may appear absent in even in healthy persons, being only visible in 60 to 90%.7,8 Its detection rate can be increased by using multiplanar imaging, contiguous sections and an anteroposterior orientation of the readout gradient.7 The presence of a bright signal is then inconsistent with the diagnosis of cranial diabetes insipidus.
There are more definite radiological features in LINH. These include characteristic diffuse thickening of the pituitary stalk, with a greater diameter exceeding 3.5 mm at the level of the median eminence of the hypothalamus.2,6 On MRI, the normal smooth tapering of the infundibular stalk is lost and there are varying degrees of asymmetry. A common finding is marked gadolinium enhancement of the stalk, extending into the lower hypothalamus. 2,6 Loss of the usual neurohypophyseal bright spot on MRI T1-weighted images is commonly reported, which has been shown to correlate closely with a loss of function of the neurohypophysis.7,8 The cases reported exhibited loss of the neurohypophyseal bright spot; but those with less than two years duration of diabetes insipidus had thickened pituitary stalk, enlarged neurohypophysis, or both, which subsequently disappeared on follow-up imaging.9 Additionally, the anterior pituitary is usually of normal size and signal intensity. There are some reported cases presenting with swelling of the whole pituitary gland and stalk with no appreciable anterior pituitary dysfunction. In our patient, non-contrast T1-weighted images revealed the absence of the usual bright signal of the posterior pituitary gland, a normal anterior pituitary and a bulky infundibulum. There was normal contrast enhancement of the anterior pituitary gland and vivid enhancement of the bulky infundibulum (Figures 1 and 2).
The absence of the posterior pituitary bright spot has been reported in other conditions. However, by coupling the clinical data with the MRI characteristics, the specific cause of central diabetes insipidus can usually be identified. In cases of neoplastic, surgical or traumatic infundibular distortion, symmetrical infundibular thickening is usually not a feature.
The loss of hyperintensity of the posterior pituitary has also been reported in pituitary involvement in Wegener’s granulomatosis.10 The pituitary gland appeared to be abnormally enlarged, while the normal hyperintensity of the posterior lobe was absent on the sagittal T1-weighted sequence. There was increased signal intensity of the anterior lobe on T2-weighted images, likely due to hemorrhagic elements within the granulomatous tissue. The contrast-enhanced studies showed central relative hypointense enhancement of the anterior lobe with preserved enhancement of the peripheral gland, and abnormal contrast enhancement extending to the hypothalamus in the reported cases.10 In lymphocytic hypophysitis, the pituitary gland exhibits homogeneous enhancement, in contradistinction to the heterogeneous enhancement seen in untreated Wegener’s granulomatosis. In the reported cases, diabetes insipidus persisted, requiring continuation of vasopressin and immunosuppressive therapy, Suggesting that the neurohypophysis was permanently damaged from an early stage of pituitary involvement.11 This was supported by the observation that the normal posterior pituitary bright spot on T1-weighted images did not reappear.
Similar peripheral enhancement around a hypointense area is a frequent finding in pituitary adenoma. However, obliteration of the high signal of the posterior lobe is not usually present in small adenomas, nor is diabetes insipidus a common clinical manifestation in adenomas of the anterior lobe. 7
Granulomatous hypophysitis can be part of systemic granulomatous conditions such as tuberculosis, sarcoidosis, histiocytosis X and syphilis; or isolated pituitary disease due to a foreign body reaction to a ruptured Rathke’s cleft cyst, mycotic infection or pituitary adenoma. If these conditions are excluded by clinical and histological examination, granulomatous hypophysitis is termed “idiopathic.”12 There is no consistently reliable radiologic finding in granulomatous hypophysitis. The lesion can be dumbbell-shaped, spherical or elliptical. Isointensity with the brain on T1-weighted image and heterogeneous high signal on T2-weighted images were noted. It may display homogeneous or heterogeneous enhancement throughout the parenchyma. Infundibular infiltration is always seen in advanced lesions, appearing as a tongue-like suprasellar extension on MRI.12 Although intrasellar tuberculomas have been reported in post-mortem examinations, they are exceptionally rare in clinical practice. In a reported case, the patient was initially diagnosed with pituitary adenoma. On MRI, T1-weighted images showed a mass isointense with gray matter, while T2-weighted images including fluid attenuated inversion recovery (FLAIR) revealed a heterogeneously hyperintense mass. The post-gadolinium contrast images showed moderate to marked homogeneous enhancement of the mass, pituitary stalk and surrounding dura. The stalk showed thickening and nodularity. The patient was initially treated with bromocriptine, but subsequently underwent transsphenoidal excision due to intolerance to the medication. The histopathologic diagnosis revealed intrasellar tuberculoma. The patient responded to anti-tuberculosis drugs clinically and radiologically.13
The definitive diagnosis can only be established by histopathologic findings. However, the effectiveness of a conservative approach in many cases, as well as the risks associated with pituitary biopsy or surgery, justify attempts to develop clinical diagnostic criteria.2 This potentially obviates the need for invasive procedures, except only when urgent decompression is required. Idiopathic central diabetes insipidus is considered when familial and secondary causes, such as granulomatous disease, trauma, tumors and cranial surgery, are excluded.
The clinical suspicion is high in our patient who is a young female, with an acute onset of diabetes insipidus and characteristic MRI findings. The differentiation from other lesions including rare tumors such as germinomas and Langerhan’s histiocytosis can be very difficult. These tumors may also present with diabetes insipidus and pituitary stalk thickening. However, in those circumstances, the radiologic findings are unlikely to regress, and may even progress rapidly in most cases.
Glucocorticoids have been reported effective in LAH and LINH. Reduction of the pituitary mass has been reported in 62.5% of patients who received pharmacologic doses of glucocorticoids, compared to 44.4% of patients who received only physiologic doses with lower cure rate of hormonal disorders.2 High dose methyprednisolone with a tapering dose schedule has been shown to improve anterior pituitary function and diabetes inspidus. 14 The effect was more favorable in patients with less than 6 months duration of disease, with improvement in MRI findings within 6 weeks to 6 months of therapy. Not all authors are in agreement; some believe that the effect of glucocorticoid is uncertain, other than when needed as replacement in patients with LYH.15
Our patient was treated with oral desmopressin with symptomatic improvement. Hyperprolactinemia was not treated as her regular menstrual cycle resumed prior to presentation, with no evidence of impaired fertility.
Lymphocytic hypophysitis encompasses a spectrum of distinct clinical syndromes and has important diagnostic and therapeutic implications, as it is usually mistaken for tumors requiring surgical management. In our patient, the clinical presentation was suggestive of LINH, despite her presentation 2 years after delivery. This, together with characteristic MRI findings and a negative screen for other causes, made LINH the most probable diagnosis. In the absence of evidence of secondary adrenocortical dysfunction, glucocorticoid therapy was not initiated. Her symptoms improved with oral desmopressin.
1. Panicker HK, Janicic N, Nguyen D et al. Presumed infundibuloneurohypophysitis: Unusual presentation in a postpartum patient. Am J Neuroradiol 2005;26(2):357-9.
2. Hashimoto K, Asaba K, Tamura K et al. A case of lymphocytic infundibuloneuro-hypophysitis associated with systemic lupus erythematosus. Endocr J 2002;49(6):605-10.
3. Crock PA. Cytosolic autoantigens in lymphocytic hypophysitis. J Clin Endocrinol Metab 1998;83(2):609-18.
4. Yamamoto M, Murakami Y, Nishiki M et al. A case of autoimmune hypophysitis associated with Graves’ disease. Endocr J 2002;49(6):583-7.
5. Caturegli P, Newschaffer C, Olivi A et al. Autoimmune hypophysitis. Endocr Rev 2005;26(5):599-614.
6. Colao A, Tavares LB, Pivonello R et al. “Autoimmunity and the Pituitary Gland.” In Endocrine Manifestations of Systemic Autoimmune Diseases: Handbook of Systemic Autoimmune Disease, edited by Sara E Walker and Luis J Jara. Elsevier, 2008;83-93.
7. Brooks BS, Gammal TE, Allison JD et al. Frequency and variation of the posterior pituitary bright signal on MR images. Am J Roentgenol 1989;153(5):1033-8.
8. Rivera JA. Lymphocytic hypophysitis: Disease spectrum and approach to diagnosis and therapy. Pituitary 2006;9(1):35-45.
9. Elster AD. Modern imaging of the pituitary. Radiology 1993;187(1):1-14.
10. Katzman GL, Langford CA, Sneller CM et al. Pituitary involvement by Wegener's granulomatosis: A report of two cases. Am J Neuroradiol 1999;20(3):519-23.
11. Imura H, Nakao K, Shimatsu A et al. Lymphocytic infundibuloneurohypophysitis as a cause of central diabetes insipidus. N Engl J Med 1993;329(10):683-9.
12. Vasile M, Marsot-Dupuch K, Kujas M et al. Idiopathic granulomatous hypophysitis: Clinical and imaging features. Neuroradiol 1997;39(1):7-11.
13. Singh S. Pituitary tuberculoma: Magnetic resonance imaging. Neurol India 2003;51(4): 548-50.
14. Kristof RA, Van Roost D, Klingmüller D et al. Lymphocytic hypophysitis: Non-invasive diagnosis and treatment by high dose methylprednisolone pulse therapy? J Neurol Neurosurg Psychiatry 1999;67(3):398-402.
15. Molitch ME, Gillam MP. Lymphocytic hypophysitis. Hormone Res 2007; 68(Suppl 5):145-50.
Articles and any other material published in the JAFES represent the work of the author(s) and should not be construed to reflect the opinions of the Editors or the Publisher.
Authors are required to accomplish, sign and submit scanned copies of the JAFES Declaration that the article represents original material that is not being considered for publication or has not been published or accepted for publication elsewhere.
Consent forms, as appropriate, have been secured for the publication of information about patients; otherwise, authors declared that all means have been exhausted for securing such consent.
The authors have signed disclosures that there are no financial or other relationships that might lead to a conflict of interest. All authors are required to submit Authorship Certifications that the manuscript has been read and approved by all authors, and that the requirements for authorship have been met by each author.