A 19-year old Filipino female was admitted due to multiple fractures on all extremities. About 7 months prior to admission, she noted onset of bone pain, initially at the pelvic and lumbosacral areas, progressing to include the arms and thighs. This was associated with fatigue, anorexia, muscle weakness and progressive weight loss about of 30%. She took analgesics and tolerated her condition.
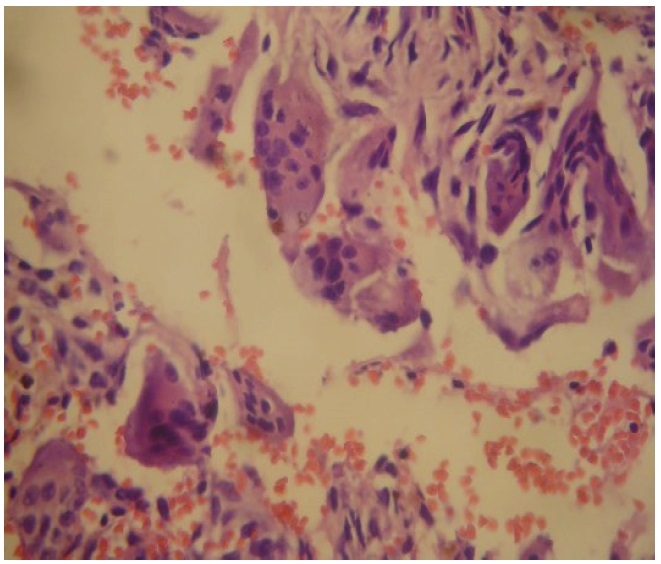
About 3 months prior to admission, she could no longer ambulate due to severe bone pains. She also noted multiple fixed hard masses on the left upper arm and leg. She was brought to a general physician for consultation following spontaneous fracture of her left humerus and left femur. She was referred to an orthopedic surgeon who worked her up as case of primary bone malignancy to rule out multiple myeloma. Bone biopsy revealed brown cell tumor/multifocal polyostotic giant cell tumor consistent with skeletal changes associated with hyperparathyroidism (Figure 1). She was subsequently transferred to our institution for further workup of the etiology of hyperparathyroidism.
Click here to download Figure 1Figure 1. Bone biopsy showing brown cell ti=umor/multifocal polyostotic cell tumor, with no findings suggestive of malignancy (H&E, x 400).
On examination, her vital signs were stable. Pertinent physical examination findings were deformities on both arms and the left thigh. These were consistent with radiologic findings of closed complete fractures on the middle third of the humerus on both arms, and the middle third of the right femur (Figure 2 and 3). She experienced severe pain even on minimal movement. She was admitted with the diagnosis of brown cell tumor with multiple lytic bone lesions secondary to primary hyperparathyroidism from a parathyroid adenoma or carcinoma.
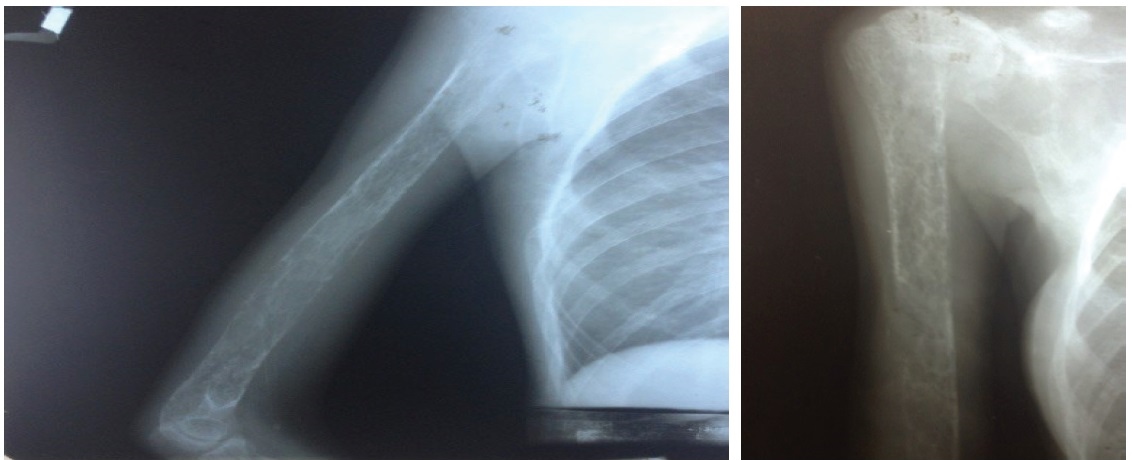
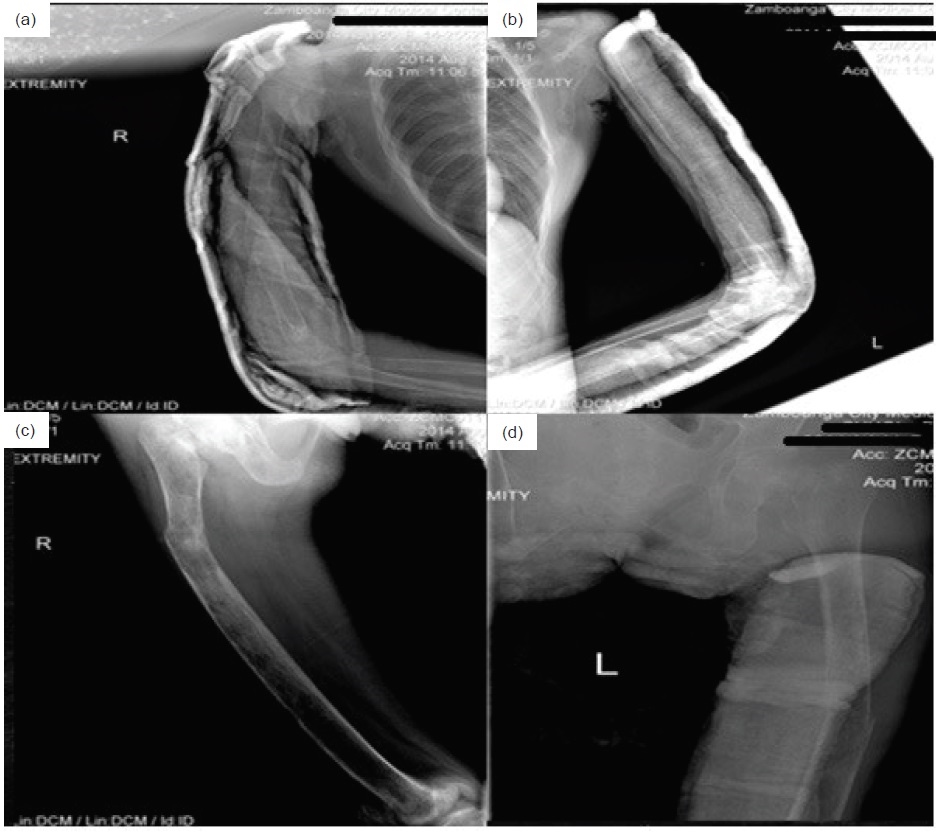
Click here to download Figure 2Figure 2. X-ray of the right humerus showed generalized decrease in bone density and subperiosteal and intracortical resorptive changes. Most of the humeral shaft demonstrated a mottled multicystic appearance with prominent trabeculations. A fracture was seen on the middle third of the humerus.
Click here to download Figure 3
Figure 3.Pathologic fractures bilateral middle 3rd humerus and right femur with angulation of distal fracture segment. Significantly generalized severe osteopenia ans subperiosteal reactions
Laboratry Work-up
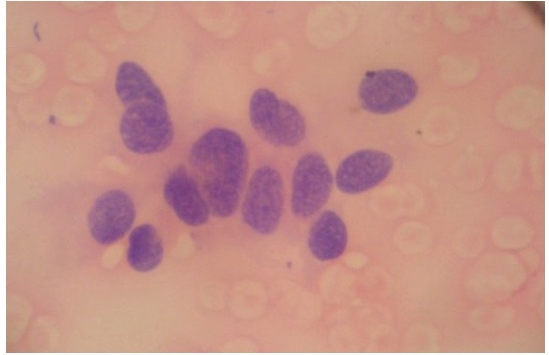
Her corrected calcium was elevated at 1361 umol/L [normal value (NV) 760 to 884 umol/L], while serum phosphorus was decreased at 221 umol/L (NV 221 to 396 umol/L ). iPTH was extremely high at 2001 pg/mL (NV 8.5 to 72.5 pg/mL). X-ray of the extremities showed osteopenia, endosteal resorptive changes and multiple pathologic fractures. Ultrasonography of the neck revealed a parathyroid mass probably an adenoma at the inferior pole of left thyroid gland measuring 2.3 cm x 1.1 cm x 1.0 cm. Sestamibi scan of the parathyroid gland was not done as this was not available in our region. An ultrasound-guided fine needle biopsy was done instead, which revealed histologic findings consistent with parathyroid carcinoma (Figure 4). Ultrasonography of the kidneys revealed bilateral nephrolithiases. Free thyroxine, triiodothyronine and thyroid-stimulating hormone levels were within normal limits.
Click here to download Figure 4Figure 4. Histologic examination of fine needle aspirate of left parathyroid. Findings were consistent with a carcinoma with spindle cell features ad clear cytoplasm, highly suggestive of parathyroid origin (H&E, x 400).
Management
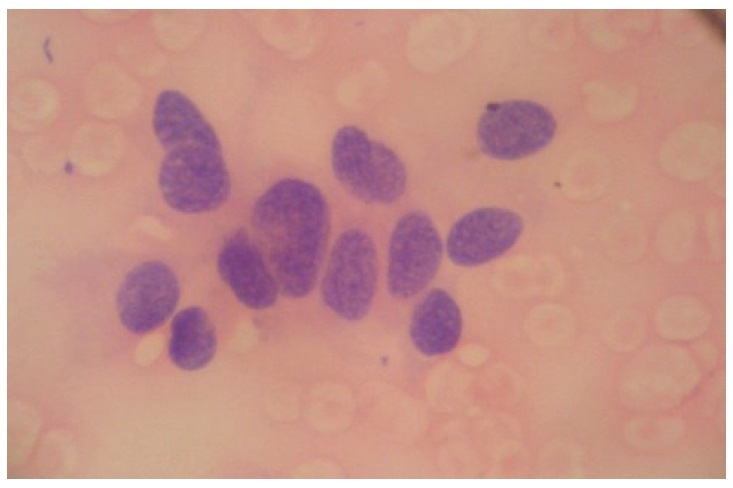
Intravenous hydration, furosemide and alendronate were given for the severe hypercalcemia. Conservative treatment with plaster reduction was done by the Orthopedics Department for the management of her multiple fractures. After 2 weeks of medical management, she underwent 3½ gland parathyroidectomy with en bloc left thyroid lobectomy. Intraoperative findings revealed a left parathyroid gland mass with poorly circumscribed borders invading the capsule and local tissues, and a well-circumscribed right parathyroid mass. Serum calcium and iPTH immediately decreased (1131 umol/L from 1255umol/L and 211.8 pg/mL from 2001 pg/mlL, respectively) one hour after the surgery. Further reduction was noted 24 hours (corrected calcium 9.2 mg/dL, iPTH 48 pg/mL) and 72 hours (serum calcium 795 umol/L, phosphorus 232umol/L) post-surgery. Histopathologic examination of the left parathyroid gland showed cytomorphologic features suspicious for neoplasm, described as areas with monotonous nuclear atypia, fibrosis and focal trabecular growth with prominent nuclear pleomorphism and scant to ample amount of granular cytoplasm (Figure 5a and b). The right parathyroid gland showed mild hypercellularity with benign epithelial cells and cystic degeneration consistent with adenoma. Immunohistochemical staining revealed negative results for TTF-1, calcitonin and chromogranin. Ki-67 study also showed low proliferation at less than 6%. These signify the absence of a neuroendocrine tumor and also indicate a slow rate of cell division of the parathyroid malignant cells.
Click here to download Figure 5Figure 5. Histologic examination of the left parathyroid gland showing hypercellularity with fibrosis, trabecular growth and areas of monotonous nuclear atypia, rather than random atypia seen in adenomas. Indisputable criteria for malignancy (i.e. capsular, vascular perinueral and thyroid invasion) were not strongly met (H&E, x 400).
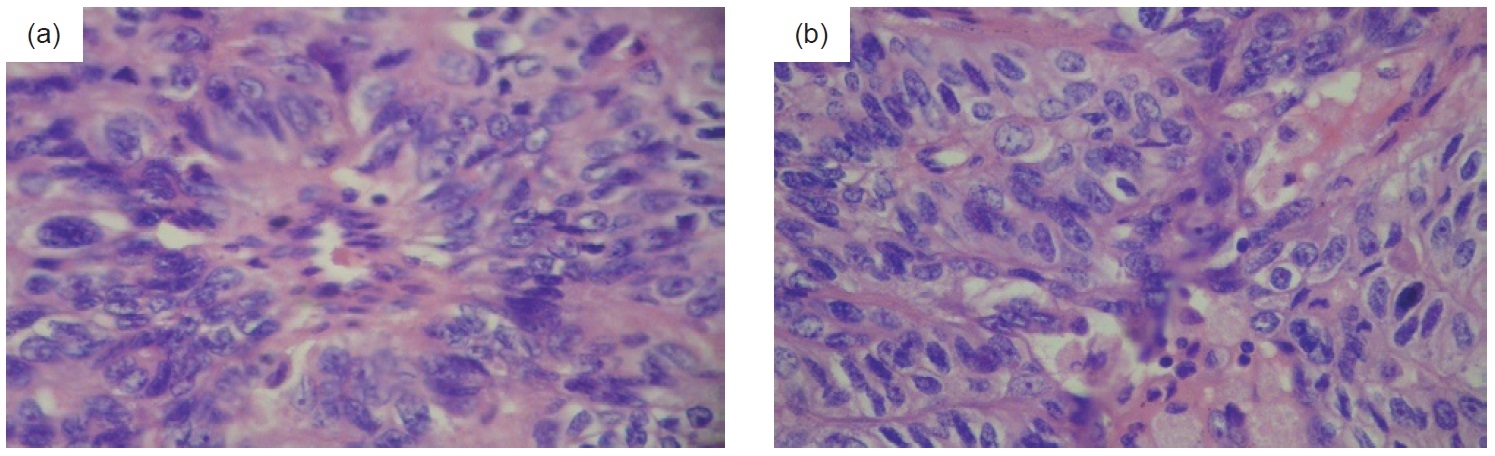
Her postoperative course was unremarkable. She was discharged 7 days after surgery with plaster reduction on all extremities, and home medications of calcium carbonate + cholecalciferol and alendronate sodium.
Outcome and Follow-up
Three months after discharge, the multiple fractures in her extremities resolved completely with no recurrence of fractures or bone pains (Figures 6 and 7). The hard masses in the left femur and tibia also disappeared.
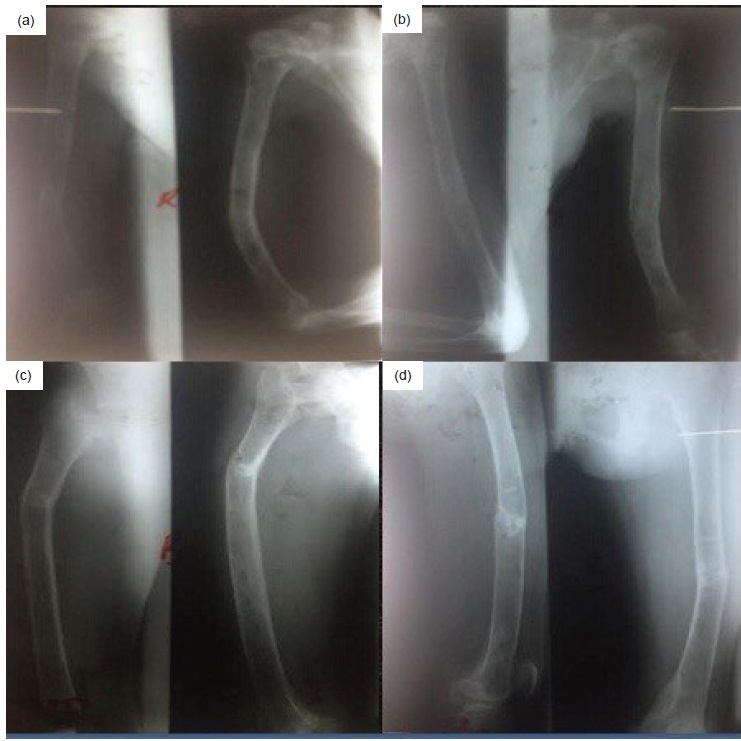
Click here to download Figure 6Figure 6. X-rays of the right humerus (a), left humerus (b,) right femur (c), and left femur (d) done 3 weeks after surgery. Callus formation was seen at the fractured segments with moderate to severe osteopenia.
Click here to download Figure 7
Figure 7. X-rays of the right humerus (a), left humerus (b), right femur (c), and left femur (d) done 3 months after surgery. Callus formation was still seen at the fractured segments, with mild osteopenia. Anterolateral bowing was alos observed in the right humerus, right femur and left femur. Anteromedial bowing was seen in the left humerus.
While parathyroid adenomas represent a common endocrine problem, parathyroid carcinomas are very rare tumors. With an estimated incidence of 0.015 per 100,000 population and an estimated prevalence of 0.005% in the United States, parathyroid cancer is one of the rarest of all human cancers.1,2 In Europe, the United States and Japan, parathyroid carcinoma has been estimated to cause hyperparathyroidism (HPT) in 0.017% to 5.2% of cases. However, many series report this entity to account for less than 1% of patients with primary HPT.1,3,4 The median age in most series is between 45 and 51 years.1 The ratio of affected women to men is 1:1, in contrast to primary HPT where there is a significant female predominance of 3 to 4:1.4
Mutation of the HRPT2 (also called CDC73) tumor suppressor gene has been recognized to play a central role in the molecular pathogenesis of parathyroid carcinoma. HRPT2 is located on chromosome 1 and encodes parafibromin, a protein whose function remains under investigation but appears to involve regulation of gene expression and inhibition of cell proliferation. Sporadic (nonfamilial) parathyroid carcinomas frequently bear HRPT2 mutations. One study reported HRPT2 mutation in 10 of 15 sporadic parathyroid cancers, while another identified the mutation in 4 of 4 carcinomas.5,6 Most mutations were somatic, implying a selective advantage that attests to their pathogenetic importance. Furthermore, because mutations outside the coding region are expected to occur but would have escaped detection, it is plausible that HRPT2 inactivation drives virtually all parathyroid cancers. Unsuspected germ-line mutations were also discovered in a substantial minority of patients who presented clinically with sporadic disease, suggesting that some of these individuals may have hyperparathyroidism-jaw tumor syndrome (HPT-JT), or a phenotypic variant.5 This recognition that family members of some patients with apparently sporadic parathyroid cancer are also at risk for parathyroid malignancy has created a new indication for genetic testing.5
The frequency of the various parathyroid lesions underlying hyperfunction is 75 to 80% from adenoma, 10-15% from primary hyperplasia, and less than 5% from parathyroid carcinoma.7 These tumors secrete parathyroid hormone producing hyperparathyroidism, which is usually severe. Parathyroid carcinoma may be suspected but it usually cannot be confirmed prior to operation.8,9 Parathyroid carcinoma tends to be localized in the inferior parathyroid glands. One study reported that the primary tumor originating in the inferior parathyroid glands was found in 15 of 19 cases involving local invasion.4
Certain clinical features may help to distinguish parathyroid carcinoma from adenoma. Parathyroid carcinoma should be suspected clinically if any of the following are present: (1) hypercalcemia greater than 14 mg/dL and serum PTH levels more than twice that of normal, (2) hypercalcemia with a palpable neck mass (3) hypercalcemia associated with unilateral vocal cord paralysis, and (4) markedly elevated serum PTH accompanying renal and skeletal disease.10,14 Our patient presented with multiple fractures on all extremities, nephrolithiasis, severe hypercalcemia and a markedly elevated serum PTH. Ultrasonographic findings also revealed a parathyroid mass at the inferior pole of the left thyroid lobe, which was later confirmed to be a carcinoma.
No effective medical therapy for parathyroid carcinoma is known. Trials of chemotherapeutic agents have been generally disappointing, with only anecdotal reports of success. This tumor is sufficiently rare that controlled trials are impossible.9 Medical therapy is primarily geared toward management of the hypercalcemia that is often quite severe. Treatment is similar to hypercalcemia due to other causes. At initial presentation and for rapid treatment of severe hypercalcemia, volume loading and diuresis with a calcium-wasting loop diuretic is the treatment of choice. The next line of therapy, used for chronic treatment, is the bisphosphonates. Hypercalcemia due to parathyroid cancer is often resistant to long-term medical management and is usually the cause of death in patients with metastatic disease.9
Surgery is the mainstay of therapy. The approach to initial surgery has evolved from an extremely aggressive procedure with ipsilateral thyroidectomy, isthmusectomy, excision of nearby strap muscles and removal of the recurrent laryngeal nerve, to a less aggressive one, involving parathyroidectomy or en-bloc resection of the parathyroid mass and any adjacent tissues that have been invaded by tumor.14
In a study of Anderson, Samaan and Vassilopoulou, the disease typically follows one of 3 courses: (1) a third of patients are cured at initial or follow-up surgery, (2) another third recur after a prolonged disease-free survival but may be cured with reoperation, and (3) the remainder experience a short and aggressive course with a high recurrence rate, even after a seemingly successful surgery. The combined 5- and 10-year survival rates varied from 50 to 70% and 13 to 35%, respectively.14
In some cases it may not be possible to differentiate parathyroid adenoma or carcinoma at the time of diagnosis or initial surgery. Local recurrence or occurrence of distant metastases at subsequent follow-up ultimately determines the correct pathological diagnosis.12 Approximately 40% to 60% of patients experience a post-surgical recurrence, typically in the range of 2 to 5 years after the initial resection.15
Severe hypercalcemia in the presence of multiple pathologic fractures, nephrolithiasis, elevated iPTH and constitutional signs and symptoms including bone pain, fatigue, anorexia, nausea, vomiting, weight loss and dehydration strongly point to a diagnosis of primary hyperparathyroidism from parathyroid carcinoma. Complete history, physical examination, a high index of suspicion and histopathologic confirmation is necessary for the diagnosis.
1. Rahbari R, Kebebew E. Parathyroid tumors. In: DeVita VT Jr, Lawrence TS, Rosenberg SA: Cancer: Principles and Practice of Oncology. 9th ed. Philadelphia, Pa: Lippincott Williams & Wilkins, 2011. 2. Hundahl SA, Fleming ID, Fremgen AM, Menck HR. Two hundred eighty-six cases of parathyroid carcinoma treated in the U.S. between 1985-1995: A National Cancer Data Base Report. The American College of Surgeons Commission on Cancer and the American Cancer Society. Cancer. 1999;86(3):538-44. 3. Fraker DL. Update on the management of parathyroid tumors. Curr Opin Oncol. 2000;12 (1):41-8. 4. Shane E. Clinical review 122: Parathyroid carcinoma. J Clin Endocrinol Metab. 2001;86 (2):485-93. 5. Shattuck TM, Välimäki S, Obara T, et al. Somatic and germ-line mutations of the HRPT2 gene in sporadic parathyroid carcinoma. N Engl J Med. 2003;349:1722-9. http://dx.doi.org/10.1056/NEJMoa31237. 6. Howell VM, Haven CJ, Kahnoski K, et al. HRPT2 mutations are associated with malignancy in sporadic parathyroid tumours. J Med Genet. 2003;40:657-63. http://dx.doi.org/10.1136/jmg.40.9.657. 7. Cotran RS, Kumar V, Collins T. Robbins Pathologic Basis of Disease. 6th ed. Philadelphia: Saunders, 1999. 8. Busaidy NL, Jimenez C, Habra MA, et al. Parathyroid carcinoma: A 22-year experience. Head Neck. 2004;26(8):716-26. http://dx.doi.org/10.1002/hed.20049. 9. Clayman GL, Gonzalez HE, El-Naggar A, Vassilopoulou-Sellin R. Parathyroid carcinoma: Evaluation and interdisciplinary management. Cancer. 2004;100(5):900-5. http://dx.doi.org/10.1002/ cncr.20089. 10. Wynne AG, van Heerden J, Carney JA, Fitzpatrick LA. Parathyroid carcinoma: Clinical and pathologic features in 43 patients. Medicine (Baltimore). 1992;71(4):197-205. 11. Nikkilä MT, Saaristo JJ, Koivula TA. Clinical and biochemical features in primary hyperparathyroidism. Surgery. 1989;105(2 Pt 1):148-53. 12. Vetto JT, Brennan MF, Woodruf J, et al. Parathyroid carcinoma: Diagnosis and clinical history. Surgery.1993;114(5):882-92. 13. Hoelting T, Weber T, Werner J, Herfarth C. Surgical treatment of parathyroid carcinoma (Review). Oncol Rep. 2001;8(4):931-4. http://dx.doi.org/10.3892/or.8.4.931. 14. Anderson BJ, Samaan NA, Vassilopoulou-Sellin R, Ordonez NG, Hickey RC. Parathyroid carcinoma: Features and difficulties in diagnosis and management. Surgery. 1983;94(6):906-15. 15. Sandelin K, Auer G, Bondeson L, Grimelius L, Farnebo LO. Prognostic factors in parathyroid cancer: A review of 95 cases. World J Surg. 1992;16(4):724-31. http://dx.doi.org/10.1007/ BF02067369.
Authors are required to accomplish, sign and submit scanned copies of the JAFES Declaration that the article represents original material that is not being considered for publication or has not been published or accepted for publication elsewhere.
Consent forms, as appropriate, have been secured for the publication of information about patients; otherwise, authors declared that all means have been exhausted for securing such consent.
The authors have signed disclosures that there are no financial or other relationships that might lead to a conflict of interest. All authors are required to submit Authorship Certifications that the manuscript has been read and approved by all authors, and that the requirements for authorship have been met by each author.