Achmad Zulfa Juniarto, MD
Department of Pediatrics, Sophia Children's Hospital, ErasmusMC
PO Box 2060, 3000 CD, Rotterdam, the Netherlands
Tel. No.: 31-47036643
Fax No.: 31-10-7036811
E-mail: a.juniarto@erasmusmc.nl; zulfa_juniarto@yahoo.com
e-ISSN 2308-118x
Printed in the Philippines
Copyright © 2012 by the JAFES
Received July 26, 2012. Accepted October 29, 2012.
Objective. Early diagnosis of congenital adrenal hyperplasia (CAH) and the need for long term treatment are of great concern to the medical community. The aim of this study was to evaluate the effectiveness of glucocorticoid treatment-monitoring based on salivary 17-hydroxyprogesterone (17-OHP) and androstenedione measurements in CAH patients in Semarang, Indonesia in comparison to patients treated in Rotterdam, the Netherlands.
Methodology. 25 out of 43 patients with CAH from Semarang, Indonesia were included in the study. For comparison, twenty CAH patients from the Sophia Children’s Hospital/ Erasmus Medical Center Rotterdam, the Netherlands were included. The effects of treatment were monitored by estimations of the steroids 17-OHP and androstenedione in saliva. Auxology and bone age determination were recorded.
Result. 17-OHP and androstenedione levels were high in a substantial number of Indonesian patients under treatment but decreased after adjustment of dosage and timing. The steroid concentrations obtained after adjustment were similar to those found under comparable circumstances in patients in Rotterdam.
Conclusion. Optimal treatment of CAH patients in Semarang Indonesia can be reached by introducing hydrocortisone treatment and adjusting dosage and timing on the basis of salivary steroid monitoring. However, in Indonesia the management of these patients is still constrained by the lack of diagnostic and therapeutic means.
Keywords: Congenital Adrenal Hyperplasia, saliva, 17-hydroxyprogesterone, androstenedione, Indonesia
Congenital Adrenal Hyperplasia (CAH is classified as a Disorder of Sex Development (DSD) which is usually diagnosed on the basis of ambiguous genitalia in the female newborn or a salt losing crisis which may occur in both males and females.1, 2 CAH infants with ambiguous genitalia are genetically female (46,XX) whereas 46,XY infants do not have a significant phenotype except for hyperpigmentation of the scrotum. This disorder occurs when one of the enzymes required for synthesis of cortisol and aldosterone in the adrenal glands is deficient. The condition results in hyperplasia of the adrenal glands and in increased secretion of steroid precursors, such as 17-hydroxyprogesterone (17-OHP) and androstenedione, leading also to elevated testosterone levels.3
Based on its clinical appearance, CAH patients can be divided into two groups, those with the classical and those with the non-classical form. The classical form is characterized by congenital insufficiency of cortisol and aldosterone synthesis with a marked increase of androgen levels. This condition will lead to salt losing and virilization of the female. The non-classical form is characterized by partial deficiency of cortisol and aldosterone synthesis resulting in late onset of mild clinical symptoms.4
The incidence of the classical form of CAH is approximately 1 in 14.000 births worldwide whereas that of the non-classical form is predicted to be much higher.5
Early diagnosis and treatment of CAH patients is essential. The need for long term treatment especially of salt losing patients is of great concern to the medical community in view of the high risk of hyponatremia, hyperkalemia, dehydration and shock with a possibly fatal result.
Treatment of CAH patients aims to provide adequate adrenal hormone substitution in order to prevent adrenal crises and to suppress excess testosterone production. The goal of therapy is to optimize growth and pubertal development.6 Hormone replacement therapy using hydrocortisone (HC) is an option that allows control of the adrenal hormone balance. The effectiveness of therapy is measured by monitoring the adrenal precursor steroids. 7 In a previous study we confirmed that determination of salivary androstenedione and 17-OHP in CAH patients is a useful alternative to the measurement of these hormones in serum.8
This is particularly relevant in Indonesia as the availability of treatment and monitoring of CAH is limited due to the scarcity of medicine, geographical distance and scattered area of patients’ dwelling, and financial issues.
Therefore, the aim of this study was to evaluate the effectiveness of glucocorticoid treatment-monitoring based on salivary 17-OHP and androstenedione measurements among Indonesian CAH patients in Semarang in comparison to CAH patients treated in Rotterdam, the Netherlands as reference. The Netherlands is a West-European country counting around 17 million inhabitants where newborns are screened within the first week of life for elevated 17-OHP levels to identify congenital adrenal hyperplasia. The prevalence of classic CAH in the Netherlands is about one in 10,000 to 18,000 newborns, of which 95% is caused by 21-hydroxylase deficiency.
This study is part of a stepwise diagnostic evaluation of a cohort of patients with DSD in Semarang Indonesia. Out of 286 patients diagnosed with disorders of sexual development between 2004 and 2010 from the Dr. Kariadi University Hospital in Semarang, Central Java, Indonesia, 43 patients were diagnosed as CAH based on phenotype and hormonal analysis. This diagnosis was confirmed by chromosomal and gene mutation analysis. Patients’ age at the time of referral ranged from 3 days to 33 years with a median of 6 years. Among the 43 CAH patients, 40 had 46,XX karyotype; two had 46,XY and one had 46, XX (99%) / 46, XY (1%) karyotype.
The latter patient presented at age 0.2 years with undescended testis and severe hypospadias. All patients with a Y chromosome were raised as males, while of the 40 CAH patients with 46, XX karyotype, 5 were raised as males and 35 as females. Thirteen patients were diagnosed at an early age (< 2 months), 23 with ages between 1.5 and 13 years while 7 were detected after 12 years of age. Five patients died during the study period because of a salt losing crisis, 10 were not treated for the following reasons: 5 patients were living as males and another 5% refused the treatment.
Twenty-eight out of 43 patients were treated and 25 patients were regularly followed up in our outpatient clinic, while 3 patients were followed up in another centre.
The effect of glucocorticoid treatment was studied in these 25 CAH patients: 2 patients received prednisone while 23 patients received HC. Patients age at treatment ranged between 0.7 - 18.7 years (see Table 1).
Patients were treated with HC at a dose of 12-15 mg/m2/day given in 3 divided doses. In 2 adult patients age 18.7 and 17.5 years old, prednisone was given at a dose of 5 mg twice a day. <
Click here to download Table 1
Table 1. Characteristics of patients
Six patients were suspected to have the salt wasting type of CAH. This diagnosis was based on a history of intercurrent illnesses during therapy. We could not establish the diagnosis as no data on urinary salt loss and renin levels were available. These patients were treated additionally with 0.05-0.2 mg/day of 9α-fludrocortisone, a synthetic mineralocorticoid.
The regular follow up every 3 months included physical examinations and measurements of blood pressure, height and weight. Instructions were given to parents and patients which included dosing schedules for medication and check of compliance. Auxological parameters, such as growth velocity and weight were also recorded. The effect of treatment was regularly monitored by assessment of salivary levels of 17-OHP and androstenedione. As much as possible an x-ray of the left hand for bone age determination was obtained once a year in prepubertal and pubertal children. In addition, we obtained auxological data in prepubertal children
The Dutch patients were included in the study to provide a reference of salivary steroid precursor concentrations in well-controlled congenital adrenal hyperplasia patients treated in a tertiary reference outpatient-clinic in a country with one of the best health-care systems worldwide. As there are no reference values available for salivary adrenal steroid precursor concentrations, this population provides the next best reference. Twenty CAH patients from the outpatient clinic of the Sophia Children’s Hospital/Erasmus MC, Rotterdam, the Netherlands were included in this study. The age of the patients ranged between 5-16 years, 13 were males and 7 females; 12 patients had a salt wasting type of CAH. All patients in this study were treated with HC, 8.8 to 17.5 mg/24h per square meter body surface area, divided in three doses per day. An additional treatment with 9α-fludrocortisone at a dose of 0.05-0.2 mg/day was given to the salt wasting patients.
Salivary concentrations of 17-OHP and androstenedione were determined using salivary ELISA Kits (DRG, Amersfoort, the Netherlands).
After a clear explanation of the procedure, the patients were asked to collect saliva by passive drooling into Sarstedtâ (Nümbrecht, Germany) polypropylene tubes or, for babies, by pipetting with a minimum volume of 1 ml before eating or tooth brushing in order to prevent blood contamination. Contamination with food debris was avoided by rinsing the mouth with water. Collection times were as follows: the first sample before the morning tablet of medication at 06.00-07.00 h, the next three samples at 12.00-13.00, 16.00-17.00 and 21.00-22.00 for two successive days. Saliva was stored frozen at -20°C until measurement in order to avoid microbial decomposition of the steroids.
Height standard deviation scores (SDS) were calculated with a growth calculator using Growth Analyzer version 2 (www.growthanalyser.org). Growth data of the Chinese population in the programme were used as reference as no data are available for the Indonesian population. Body mass index (BMI) was calculated as weight (kilograms)/height (meters)2.
Bone age was assessed by x-ray of the left hand using the Greulich and Pyle method.9
The patients or their parents gave informed consent to participate in this study, which was approved by the ethics committee of both institutions.
Click here to download Figure 1
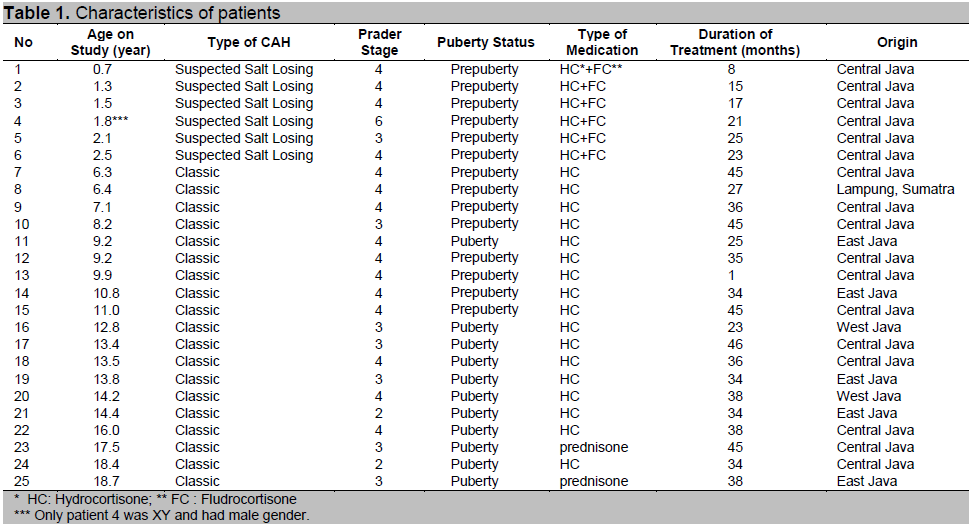
Figure 1. Salivary 17-hydroxyprogesterone and androstenedione diurnal rhythms. Circadian rhythms of 17-hydroxyprogesterone and androstenedione of 25 CAH patients on two subsequent days (full lines). Their levels were significantly correlated. In the morning, the concentrations of these steroids were high and then decreased gradually until the lowest values were found in the evening. A significant decline of hormone levels occurred after adjustment of dosage and time of administration of the medication (p = 0.01 and 0.04 respectively for 17-OHP and androstenedione) (broken lines). Figure shows means and SEM. Reference ranges for 17-hydroxyprogesterone and androstenedione are below 225 and 250 pmol/l, respectively.
After a period of steroid treatment varying between 1 - 46 months (median of 34 months), 17-OHP and androstenedione levels in saliva were measured four times a day prior to HC administration for two consecutive days for each patient to obtain the diurnal rhythm.
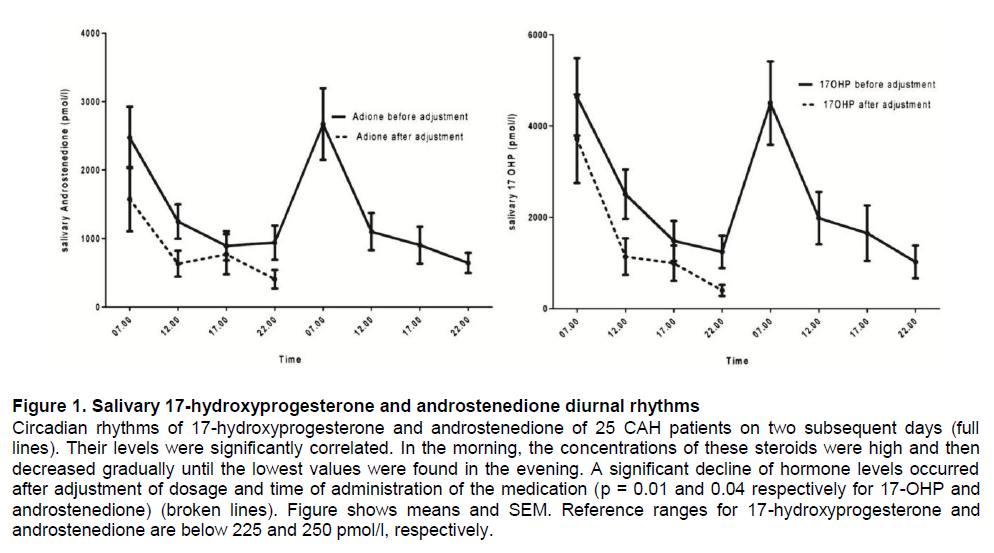
Results are shown in figure 1, indicating high levels in a substantial number of patients. Therefore the dosage of HC and particularly the timing were adjusted. Based on these results, additional saliva samples were obtained after 1 month of adjusted treatment and just prior to the administration of HC for one day. Saliva steroid precursor concentrations decreased after treatment was adjusted (Figure 1). A clear correlation between saliva levels of 17-OHP and androstenedione was confirmed (r = 0.88).The morning saliva steroid concentrations of the patients in Semarang obtained after adjustment of dosage and timing were compared to steroid concentrations in saliva of CAH patients in Rotterdam obtained under comparable circumstances. As shown in Figure 2, the concentrations were comparable for most patients.
With regard to bone age examinations, 11 patients already stopped growing before start of therapy, 5 patients were examined only once, and in five patients no data were available. In 4 patients, the progression of bone maturation could be analyzed and all were found to have advanced bone age; The progression of maturation decreased in 3 patients after 45, 45, 35 months of therapy respectively.
Of the 16 patients who were still experiencing growth, 8 had improvement in height SDS during therapy, 3 experienced a decrease, 2 were unchanged and the other 3 could not be assessed because no data were available (Table 2).
Lifelong use of glucocorticoids is the therapy of choice in patients with CAH. Dose adjustments to avoid the adverse consequences of inadequate treatment and overtreatment are based on rigorous assessment of biochemical parameters.
This study shows that the result of treatment of CAH patients in Semarang, Indonesia is comparable to that in a Dutch endocrine centre. Both centres utilized salivary steroid measurements to monitor and adjust HC treatment.
Click here to download Figure 2
Figure 2. Salivary steroid levels in CAH patients in Semarang and Rotterdam Comparison of the levels of 17-hydroxyprogesterone and androstenedione in saliva samples collected from CAH patient in Semarang (after treatment adjustment) and Rotterdam showing similar concentrations both in prepubertal and pubertal patients. (Prepubertal age: <12 years, pubertal age : ≥12 years) Reference ranges for 17-hydroxyprogesterone and androstenedione are below 225 and 250 pmol/l, respectively.
Click here to download Table2
Table 2. Development of height SDS during period of HC treatment of CAH patients
The purpose of early treatment of CAH is to suppress the excessive adrenal androgen secretion without disrupting growth and the development of puberty. To achieve this, the glucocorticoid dosage should not exceed the physiological secretion of cortisol (6 mg/m2/day) which is equivalent to a HC intake of 8-10 mg/m2/day.
The deficiency of the adrenal enzyme 21-hydroxylase leads to markedly increased concentrations of steroid precursors from the adrenal gland. Thus dosing glucocorticoids at slightly supra-physiological levels will suppress the increased adrenal androgen production.10 This can be achieved with an intake of HC 10-15 mg/m2/day
While our results indicate that suppression can be achieved, adjustment of dosage as well as time of dosing was necessary to reach an acceptable degree of suppression that was comparable to results in another clinic. When the medication was taken at defined hours (07-13-18 hr), the profile of hormones showed a steep increase in the morning and a gradual decline in the evening. This is because intake of HC in the early evening (18 hr) is insufficient to cover the corticosteroid requirement until the next morning. Therefore, we advised to administer the medication regularly every 8 hours at 06-14-22 hr to improve the hormonal profile.
HC is preferred over prednisone or dexamethasone in infants and children for the treatment of CAH because it is well tolerated, leads to less side-effects because of its shorter half-life and it is inexpensive.6 Most importantly, it will not inhibit linear growth provided it is given in the correct dosage. In prepubertal and pubertal children, treatment with long acting glucocorticoids such as prednisone or dexamethasone is no longer used because of the side effects of suppressing linear growth. Slow release preparations of HC accommodating the circadian rhythm of cortisol in the body are not yet available.11 Thus, specifically in infants and young children, HC in a dosage adapted to body surface is the drug of choice.
Whereas the short half life of about 8 hours may allow a dosing schedule of 3 times a day, this dosing schedule may be insufficient especially in infants and young children to cover their overnight requirement. As a result, the HC does not last until the morning explaining high levels of 17-OHP and androstenedione in the morning samples of our patients, whereas the levels of both hormones decline appropriately during the day following morning and noon administration of HC. In contrast, in patients with compliance problems levels remain high throughout the day.
Monitoring HC therapy of patients with CAH, using saliva to measure steroid hormones is used extensively.7, 10 The main advantage of using saliva is the non-invasive way of collecting samples. In fact, steroid levels in saliva reflect the physiologically active fraction of steroid hormones. 6,12 Moreover, saliva collection is very patient friendly as it may be done at home at designated times. Steroids in saliva are stable at room temperature allowing to send samples by regular mail to the laboratory for measurement.
Because single sample monitoring provides a picture of adrenal hormonal production at only one point in time and does not take into account the circadian rhythm of steroid secretion, some experts favour checking the hormonal profile during 24 hours by collecting blood or saliva samples several times during the day. Drawing a blood sample is bothersome to the patient, and is often logistically not feasible.10 In several studies, steroid levels in saliva and serum have been found to have a high correlation.10,13
During periods of rapid growth such as during infancy and puberty the extent to which one should relax standards for 17-OHP, androstenedione and testosterone levels are not fully determined.14 Many of our patients (12 out of 25) were in the age range of 8-15 yr and were in (early) puberty. During this age period, it is difficult to maintain androgen suppression with HC treatment because of compliance problems as a result of pubertal behaviour and the changing endocrine milieu during puberty.15 Growth hormone and IGF 1 levels which are elevated during this period stimulate the activity of 17-hydroxylase/17,20-lyase resulting in increased adrenal androgen biosynthesis.16 Similarly in 7 out of 25 patients who started therapy during infancy HC dosing was notably difficult because of the rapid change of bodily proportions. Therapeutic efficacy was difficult to evaluate because most of our Indonesian patients were diagnosed and started therapy at a late age with advanced bone age which was not compatible with their chronological age. Due to premature epiphyseal closure, many of them did not achieve the optimal height: their average height amounted to -2.42 (±1.39) SDS.
Among the patients who were still experiencing growth, data are insufficient to draw firm conclusions. Education of patients and parents and regular monitoring of auxological parameters and saliva steroid levels will lead to further improvement of height potential. It is well recognized that the age of diagnosis, the type of CYP21 mutation and the response to a certain dose of HC in view of individual variation of drug metabolism all are relevant factors contributing to the final outcome of HC treatment.
Many patients on treatment live in remote areas. To anticipate the situation when a patient is suffering from intercurrent illness during which there is a need to increase HC dose, we offer them a reference letter that serves as information for the local doctor.
Availability of HC is very urgent because it is life-saving and it reduces morbidity not only in CAH patients but also in patients with congenital or acquired ACTH deficiency. It is believed that there are many underreported or unrecognized cases in Indonesia.
The diagnosis of CAH can be established in most of the patients with great certainty. However in Indonesia its management is still constrained by the lack of diagnostic and therapeutic means. The drug of choice in infants, young children and adolescents is HC, which is lifesaving. HC, declared an essential drug by the WHO, should be made widely available throughout Indonesia. With regular follow up HC treatment will enable a favourable outcome of patients with CAH. Saliva samples can now be collected at home and sent by regular mail to the laboratory, and overcomes a major practical monitoring problem. Collaboration with the local health provider for auxology measurements and monitoring of the health status of patients should be well promoted and will be a major step forward to optimize treatment.
This study was supported in part by a research grant from Diponegoro University (PNBP). We gratefully acknowledge the collaboration of the staff of Center for Biomedical Research (Cebior) of the Kariadi Hospital and the assistance of Ronald van der Wal, Endocrine Laboratory of the Department of Internal Medicine, ErasmusMC Rotterdam, the Netherlands.
1. Lee PA, Houk CP, Ahmed SF, Hughes IA. Consensus statement on management of intersex disorders. International Consensus Conference on Intersex. Pediatrics. 2006;118(2):e488-500.
2. Hindmarsh PC. Management of the child with congenital adrenal hyperplasia. Best Pract Res Clin Endocrinol Metab. 2009;23(2):193-208.
3. Stikkelbroeck MM, Otten BJ. [Congenital adrenal hyperplasia: clinical aspects and neonatal screening]. Ned Tijdschr Geneeskd. 2002;146(6):246-9.
4. Riepe FG, Sippell WG. Recent advances in diagnosis, treatment, and outcome of congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Rev Endocr Metab Disord. 2007;8(4):349-63.
5. Bonfig W, Pozza SB, Schmidt H, Pagel P, Knorr D, Schwarz HP. Hydrocortisone dosing during puberty in patients with classical congenital adrenal hyperplasia: an evidence-based recommendation. J Clin Endocrinol Metab. 2009;94(10):3882-8.
6. Groschl M, Rauh M, Dorr HG. Cortisol and 17-hydroxyprogesterone kinetics in saliva after oral administration of hydrocortisone in children and young adolescents with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J Clin Endocrinol Metab. 2002;87(3):1200-4.
7. Deutschbein T, Unger N, Hauffa BP, Schaaf K, Mann K, Petersenn S. Monitoring medical treatment in adolescents and young adults with congenital adrenal hyperplasia: Utility of salivary 17alpha-hydroxyprogesterone day profiles. Exp Clin Endocrinol Diabetes. 2011;119(3):131-8.
8. Juniarto AZ, Goossens K, Setyawati BA, Drop SL, de Jong FH, Faradz SM. Correlation between androstenedione and 17-hydroxyprogesterone levels in the saliva and plasma of patients with congenital adrenal hyperplasia. Singapore Med J. 2011;52(11):810-3.
9. Buken B, Safak AA, Yazici B, Buken E, Mayda AS. Is the assessment of bone age by the Greulich-Pyle method reliable at forensic age estimation for Turkish children? Forensic Sci Int. 2007;173(2-3):146-53.
10. Dauber A, Kellogg M, Majzoub JA. Monitoring of therapy in congenital adrenal hyperplasia. Clin Chem. 2010;56(8):1245-51.
11. Loechner KJ, McLaughlin JT, Calikoglu AS. Alternative strategies for the treatment of classical congenital adrenal hyperplasia: Pitfalls and promises. Int J Pediatr Endocrinol. 2010;2010:670960.
12. Woodside DB, Winter K, Fisman S. Salivary cortisol in children: correlations with serum values and effect of psychotropic drug administration. Can J Psychiatry. 1991;36(10):746-8.
13. Shimon I, Kaiserman I, Sack J. Home monitoring of 17 alpha-hydroxyprogesterone levels by filter paper blood spots in patients with 21-hydroxylase deficiency. Horm Res. 1995;44(6):247-52.
14. Speiser PW. Growth and development: congenital adrenal hyperplasia-glucocorticoids and height. Nat Rev Endocrinol. 2010;6(1):14-5.
15. Charmandari E, Hindmarsh PC, Johnston A, Brook CG. Congenital adrenal hyperplasia due to 21-hydroxylase deficiency: alterations in cortisol pharmacokinetics at puberty. J Clin Endocrinol Metab. 2001;86(6):2701-8.
16. Balducci R, Toscano V, Larizza D, Mangiantini A, Galasso C, Municchi G, et al. Effects of long-term growth hormone therapy on adrenal steroidogenesis in Turner syndrome. Horm Res. 1998;49(5):210-5.