De La Chapelle, also known as XX Male syndrome is a rare cause of male infertility.[1] Testis Determining Factor (TDF), located on the short arm of the Y chromosome, is responsible for testicular development in males. Sertoli cells secrete Müllerian Inhibiting Factor which is responsible for the agenesis of Müllerian structures. That action, along with Leydig cells that secrete testosterone, ultimately lead to male internal genitalia development. The presence of the SRY gene on X chromosome is responsible for the male phenotype in the majority of XX males, but a few of these males do not bear this chromosome. The role of certain key genes that could be implicated in abnormal sexual differentiation is known, but the complexity and heterogeneous nature of this syndrome leaves many questions unanswered. The basis of therapy is testosterone supplementation at an early stage. The objective of the case report is to highlight the importance of karyotyping during the work up of infertility in males, if azoospermia is seen on semen analysis.
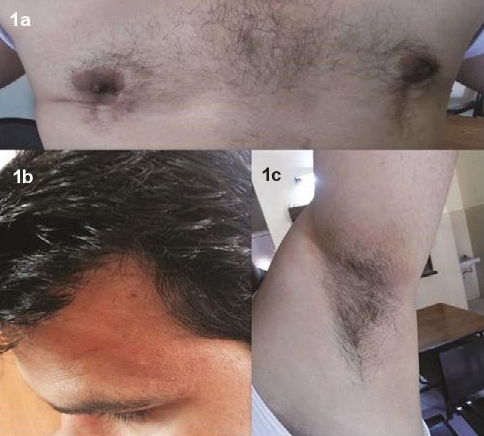
CASEA 25-year-old Indian man, nonsmoker, occasional alcoholic beverage drinker, vegetarian by diet, married for the last 2 years, presented to Endocrine OPD with complaint of inability to father a child. He had history of bilateral mastectomy a year back for gynaecomastia (Figure 1a). There were no previous records available about the work up of gynaecomastia. According to the patient, he underwent mastectomy for cosmetic purposes after his marriage. He was born as a result of nonconsanguineous marriage by full term normal vaginal delivery and cried immediately after birth. His developmental milestones were normal according to age. His height was 170 cm, weight 85 kg (BMI-29.4 kg/m2) and arm span 173 cm. His facial, axillary and pubic hair were normal in density and distribution (Figures 1b, 1c). The stretched penile length was 7 cm and testicular volume was 3 ml each (Figure 2). Respiratory, CVS, CNS, and abdominal examinations were normal.
Click here to download Figure 1Figure 1.Patient characteristics.
Click here to download Figure 2
Figure 2.Stretched penile length and testicular volumes of patient.

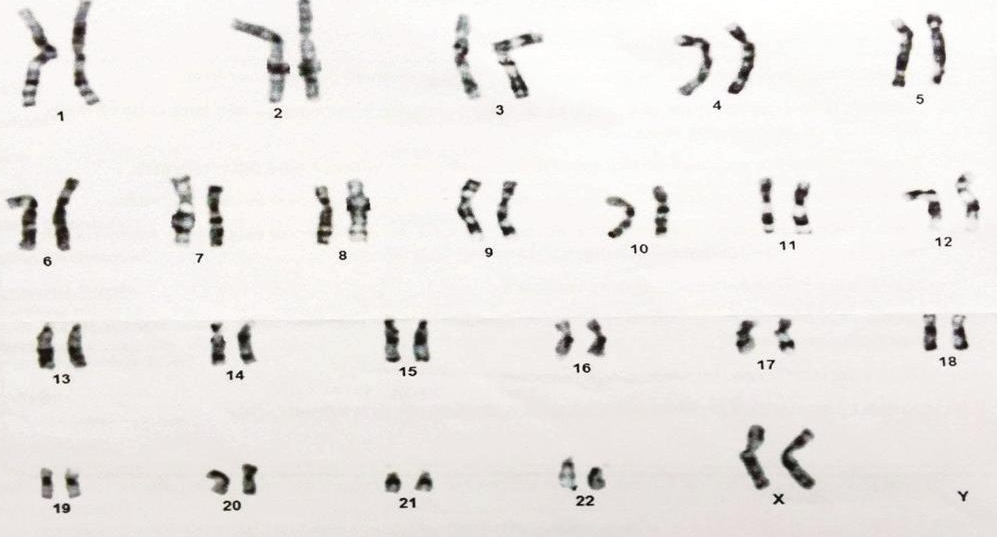
Ultrasonography and CT abdomen were normal and failed to find the uterus and adnexa. Azoospermia was reported on seminal fluid analysis. Serum luteinizing hormone (LH) and follicle stimulating hormone (FSH) were at 10.16 and 13.69 mIU/ml respectively (normal 2– 9.6 mIU/ml and 1.2–5.0 mIU/ml respectively). Serum testosterone concentration was 180.32 ng/dl (normal ranges are 270–1070 ng/dl), serum prolactin concentration was 6.32 ng/ml (normal ranges are 2.5– 17 ng/ml). Karyotyping revealed a female pattern i.e., 46,XX (Figure 3). Fluorescent in situ hybridization (FISH) to determine presence of SRY gene on X chromosome of patient was planned but patient could not get it done due to financial constraints.
Click here to download Figure 3Figure 3.Karyotyping of patient.
The authors committed to the Helsinki Convention at all stages of the investigation. An informed consent form was taken from the patient.
Infertility has traditionally been defined as the inability to conceive after 12 months of unprotected sexual intercourse. Infertility can be attributed primarily to male factors in 25% of couples and female factors in 58% of couples and is unexplained in about 17% of couples. Out of 25% of male factor infertility, primary hypogonadism accounts for 30-40% of cases, secondary hypogonadism 2% of cases, sperm transport disorders 10-20% of cases and unknown in 20-50% of cases.
Male infertility can be classified as disorders of chromosomal sex development (most common being 47XXY; Klinefelter syndrome), 46,XY disorders of sex development which include disorders of testicular development or disorders of androgen synthesis/action, primary testicular disease (uncorrected cryptorchidism, cancer chemotherapy, trauma, infectious orchitis, torsion etc.), secondary hypogonadism or hypothalamo-pituitary disease, disorders of sperm transport and aging.
Human males with a 46,XX karyotype are infertile. The incidence in newborn males is around 1 in 20,000. This syndrome accounts for 2% of cases of male infertility. Most cases are sporadic.[1] According to a proposed revised nomenclature (the Lawson Wilkins Pediatric Endocrine Society and the European Society for Pediatric Endocrinology, 2006), the diagnosis of XX male or XX sex reversal is renamed as 46,XX testicular disorder of sex development.[2] Clinical presentation depends on the presence of the SRY gene. The majority are SRY positive cases, which present as normal men after puberty with normal pubic hair and penile size but with small testes, gynaecomastia and azoospermia-related sterility. The testes size of our patient was 3 ml which was in prepubertal range as per Tanners staging. Similar presentation was seen in our patient which probably makes our patient SRY positive.
SRY-negative cases present at birth with features such as hypospadias and cryptorchidism. All males with this phenotype genotype mismatch are azoospermic, due to the absence of long arm of Y chromosome containing Azoospermia Factor gene (AZF), which is responsible for normal spermatogenesis. Studies have suggested that Y chromosomal genes such as the AZF are not only crucial for spermatogenesis but also pivotal for the maintenance of normal somatic cell function.[3]
Majority of these patients have Y chromosomal material along with SRY gene which is responsible for normal testicular development. It has been suggested that an unequal Y-to-X interchange occurs during paternal meiosis,[4] which was corroborated by more recent studies.[5] Less common causes for XX males who are SRY negative include autosomal or X chromosome gene mutations, which are responsible for testicular determination in absence of TDF, and undetermined mosaicism in Y bearing cell line.[6]-[7]
Reports from a Mexican family having two siblings without genital ambiguity were found to be SRY negative, which was suggestive of the possibility that inherent loss of function mutation of the gene participating in sex determining cascade could result in normal male sexual differentiation in absence of normal SRY gene.[8] Although incomplete masculinization is a result of the absence of Y DNA, exceptions could occur. Another possible reason for the presence of male phenotype is the influence of X inactivation on a downstream gene on the X chromosome.[9] Lastly, it has been hypothesized that this phenotype genotype mismatch is due to a defect in X-linked or autosomal sex-determining gene.[10]
Although FISH, the gold standard to determine presence of the SRY gene on X chromosome, was not done, the male phenotype and presence of gynaecomastia, azoospermia, male infertility with chromosomal pattern of XX led us to our diagnosis of 46,XX testicular disorder of sex development. Differentials of this syndrome include Klinefelter syndrome and 46,XX ovotesticular DSD. Klinefelter syndrome was ruled out as Y chromosome was not identified. 46,XX ovotesticular DSD was unlikely because of characteristic findings and no evidence of Müllerian structures on ultrasonography and CT abdomen
As most of the males develop normal male phenotype, they are raised as males. Those who fail to develop adequate secondary sexual characteristics are given testosterone replacement which improves libido and overall sexual activity, increases energy, lean muscle mass and bone density and decreases fat mass. Patients remain infertile and the only option available is by obtaining donor sperm and subsequent IUI or child adoption.
After the disclosure of the diagnosis, our patient went into denial and was not ready to accept the diagnosis. With counseling, he eventually came to terms with the fact that he cannot father a child. So, infertility being a stressful event for the couple seeking treatment, psychosocial counselling is helpful for them.
The aim of reviewing the current literature is to highlight the value of karyotyping in all males with congenital azoospermia or severe oligospermia who present for evaluation of infertility, since the male phenotype does not always guarantee the presence of Y sequence in the genome.
We conclude that owing to the rarity of this syndrome, it is easy to miss it in the differential diagnosis of phenotypically normal males with complete azoospermia. Without proper karyotyping, these patients would be subjected to financial and psychological constraints of unwanted invasive procedures. Once diagnosed, long term androgen therapy and counseling with a cooperative interdisciplinary approach would be required.
Ethical ConsiderationPatient consent form has been procured prior to the case report study.
Statement of AuthorshipAll authors have given approval to the final version submitted.
Author DisclosureAll the authors have declared no conflict of interest to the work carried out in this paper.
Funding SourceNone.
[1] De la Chapelle A. The etiology of maleness in XX men. Hum Genet.1981;58(1):105-16. DOI.