Pseudohypoparathyroidism (PHP) is a rare heterogeneous disorder. It is divided into Type 1 PHP, Type 2 PHP and pseudo-pseudohypoparathyroidism on the basis of presenting signs and investigations. Type 1 PHP presents with elevated PTH in the presence of hypocalcemia, hyperphosphatemia along with blunted phosphaturic and cAMP response to exogenous PTH infusion.[1] Type 1 PHP is further subdivided into subcategories PHP 1a, 1b and 1c. Types PHP 1a and 1c both have associated features of Albright’s Hereditary Osteodystrophy (AHO) characterized by heterogeneous clinical findings such as brachydactyly, rounded face, short stature, central obesity, subcutaneous ossifications and variable degrees of mental retardation.[2][3] Type PHP 1b can be easily differentiated clinically from PHP 1a and 1c by the absence of AHO in the former subtype. Patients with PHP 1a and 1c also have resistance to other hormones acting through G protein coupled receptors like TSH, GHRH and gonadotropins. Differentiation between PHP 1a and PHP 1c requires genetic analysis of erythrocyte Gsα activity which is reduced in PHP 1a while it is normal in PHP 1c.[1] Type 2 PHP is characterized by resistance to PTH in the absence of AHO along with resistance to other hormones, while pseudo-PHP is characterized by the presence of AHO and the absence of any hormone resistance.[1]
Turner syndrome is characterized by the absence of the complete or a part of a normal sex chromosome in females and associated features including congenital lymphedema, short stature and gonadal dysgenesis.[4] Approximately half of the patients with Turner syndrome have monosomy X while the rest have other abnormalities.[4]
Since both these disorders usually present with short stature and delayed puberty, the diagnosis and management can be challenging if both disorders are present in the same patient. We describe a case of an Asian Indian female who presented with short stature and delayed puberty along with other physical features suggestive of overlapping of PHP and Turner’s syndrome.
CASEA 15-year-old Asian Indian girl presented in the Endocrinology outpatient department with chief complaints of short stature and delayed puberty. She also complained of small 3rd, 4th, 5th toes and fingers. She was born of a non-consanguineous marriage with full term normal vaginal delivery. She attained all developmental and social milestones at the same age as her peers. At the age of 8 years, her parents noticed growth retardation along with abnormality in the shape of her toes and fingers. There was no significant history of repeated childhood hospitalizations, seizures, obesity, mental subnormality, head trauma, abnormal calcifications or edema of extremities, delayed dentition and enamel hypoplasia.
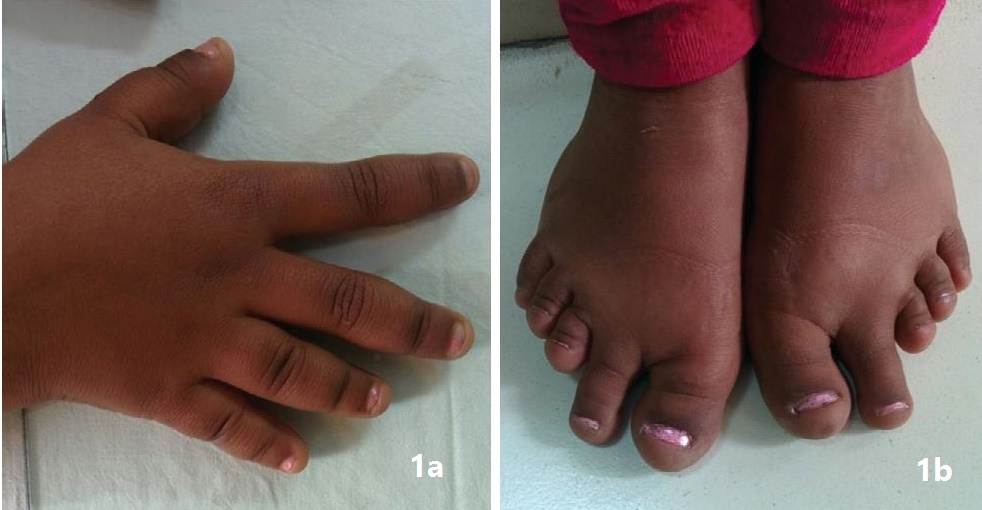
Clinical evaluation revealed a round small face, brachydactyly (shortening of 3rd, 4th, 5th fingers and toes) [Figure 2a,b] and cubitus valgus. Anthropometry showed proportionate short stature (Figure 1) with a height of 127 cm (< 3rd percentile, Standard deviation score: -4.44, mean parental height: 156 cm), arm span of 120 cm while her upper segment: lower segment ratio was 1.01. Her body mass index (BMI) was 14.17 kg/m2. Her Tanner’s staging was pre-pubertal with no axillary and pubic hair and no evidence of thelarche. Her blood pressure was normal in all four limbs. There was no evidence of any subcutaneous ossification. Her thyroid gland was not palpable. Her systemic examination was unremarkable.
Click here to download Figure 1Figure 1.Round face with typical AHO facies with short stature seen in the elder sister (right) suffering from pseudohypoparathyroidism with Turner syndrome looking shorter than 12 years. On the left is her younger sister.

Click here to download Figure 2
Figure 2a.Right hand showing brachydactyly; 2b. Showing shortening of 3rd, 4th & 5th toes bilaterally.
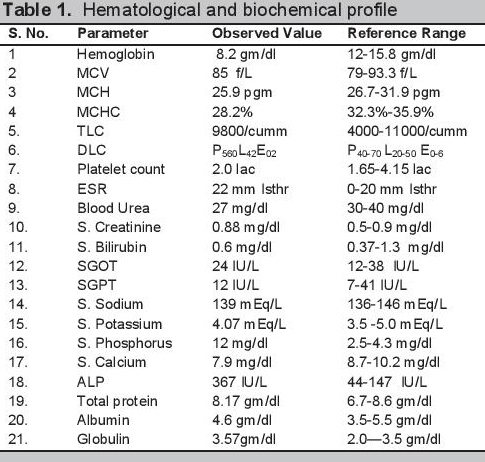
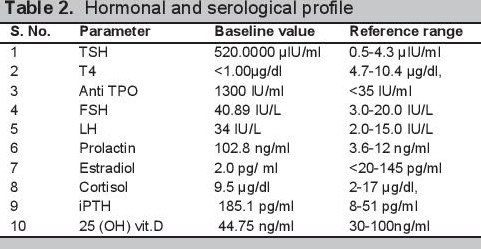
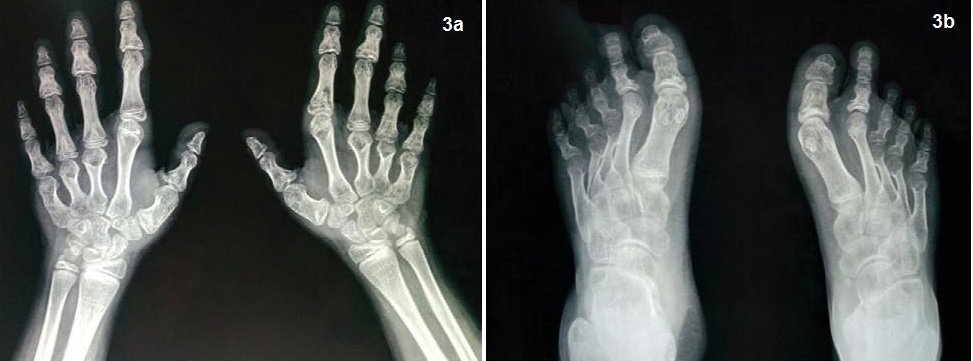
Biochemical evaluation revealed microcytic hypochromic anemia, hypocalcemia and hyperphosphatemia (Table 1). The hormonal investigations were done through chemiluminescence immunoassay by Abbott ARCHITECT i1000sr immunoassay analyzer USA using fasting plasma sample which revealed elevated parathormone level, autoimmune hypothyroidism, hypergonadotropic hypogonadism along with raised prolactin (Table 2). Roentgenographical assessment revealed bilateral short 3rd, 4th and 5th metacarpals and metatarsals with positive Archibald sign [Figure 3a,b] while the rest of the skeletal survey was normal. Ultrasonography of the abdomen showed streak ovaries along with pre-pubertal uterus. Her karyotype showing 45 (X,O) confirmed the diagnosis of Turner syndrome. Electrocardiography, 2D echocardiography and pure tone audiometry were unremarkable while thyroid ultrasonography revealed hypo-echogenicity of the thyroid gland.
Click here to download Table 1Table 1.Hematological and biochemical profile
Click here to download Table 2
Table 2.Hormonal and serological profile
Click here to download Figure 3
Figure 3.X-ray both hands and feet showing short 3rd, 4th and 5th metacarpals; 3b. Metatarsals in both limbs with positive Archibald sign.
On the basis of clinical history, examination and investigations, the diagnosis of Turner syndrome along with PHP with autoimmune hypothyroidism and nutritional deficiency anemia was made. The patient was started on treatment with thyroxine replacement, calcium, estradiol, haematinics and 1,25(OH)2 vitamin D and follow up advice after 2 months.
After 2 months of therapy, the patient showed significant clinical and biochemical improvement. On follow up visit after 2 month, her hemoglobin was 11.2 gm/dl. Her thyroid functions were improved with TSH 6.4329 μIU/m and T4 5.85 μg/dl along with normalization of serum prolactin (S. Prolactin: 5.5 ng/ml). The patient became normocalcemic and normophosphatemic with serum calcium 9.2 mg/dl and serum phosphorus 6.47 mg/dl and her parathormone level was 74.2 pg/ml.
PHP is a heterogeneous disorder with a variety of manifestations. The prevalence of the disorder is about 0.79 per 100,000.[1] It was the first hormone resistance syndrome ever described. The first description of pseudohypoparathyroidism with Albright hereditary osteodystrophy was made in 1942 by Fuller Albright.[5] In his original work, Albright described a person with hypocalcemia and hyperphosphatemia with normal renal function who had no calcemic and phosphaturic response to bovine parathyroid extract as compared to hypoparathyroid patients, hence the concept of hormone resistance came into being.[5] Later on, with the discovery of the PTH receptor and its mechanism of action, the basic pathophysiology of the disorder was described and the syndrome was further subdivided into various types.[1] The primary pathological mechanism was described as a GNAS mutation which leads to defective activity of Gsα (stimulatory subunit of G protein coupled receptors).[1] PHP has various manifestations due to the involvement of maternal imprinting of the genes.[1] PHP 1a is characterized by parathormone resistance (elevated PTH in spite of hypocalcemia and hyperphosphatemia; poor cAMP and phosphaturic response to exogenous PTH administration) along with features of Albright’s hereditary osteodystrophy (AHO), characterized by brachydactyly, rounded face, short stature, central obesity, subcutaneous calcifications in conjunction with variable mental retardation.[1] Together with variable PTH resistance, resistance to other hormones operating through G protein coupled receptors can also be present in PHP.[1] The patient in the present case had short stature, a round face, brachydactyly [Figures 1 and 2a,b] and elevated PTH in spite of hypocalcemia and hyperphosphatemia (Table 1). Along with these features, the patient also had delayed puberty and no development of secondary sexual characteristics due to hypergonadotropic hypogonadism. Her skeletal survey showed specific changes associated with AHO such as short 3rd, 4th and 5th metacarpals with short distal phalange of her thumb but the absence of heterotropic calcification.
Turner syndrome is characterized by a combination of typical physical features in phenotypic females and a complete or partial absence of the second sex chromosome, with or without cell line mosaicism.[6] Short stature is the most common physical finding in the patient with Turner syndrome and is caused by haploinsufficiency of the SHOX gene.[7] Other physical features of Turner syndrome are short fourth metacarpal, shield chest, cubitus valgus, high arched narrow palate, epicanthal folds, genu valgum, etc. Together with these features, patients with Turner syndrome are prone to develop hypertension, diabetes mellitus, coarctation of the aorta, aortic dissection, inflammatory bowel disease, hypothyroidism and many autoimmune diseases.[6][7] Our patient presented with short stature, delayed puberty, cubitus valgus and brachydactyly. On investigation, she was found to have hypergonadotropic hypogonadism and autoimmune hypothyroidism. Her karyotype revealed X chromosome haplo-insufficiency.
So far in medical literature, only one case of pseudohypoparathyroidism 1a with Turner syndrome has been described.[8] Another case of pseudopseudohypoparathyroidism with Turner syndrome and one case with pseudohypohyperparathyroidism have been reported.[9][10] The only case of pseudohypoparathyroidism with Hashimoto’s thyroiditis with Turner syndrome was described by Wen Heng-Zeng et al., in 2014 in which they described the case of a 16-year-old female presenting with short stature. On examination, round chubby face, short neck, short stature and obesity were observed. Biochemical investigations revealed elevated parathormone, thyroid stimulating hormone (TSH), prolactin, follicle stimulating hormone (FSH), antithyroid peroxidase antibody and anti-thyroglobulin antibody levels. The radiological examination showed delayed bone age, immature uterus along with shortening of metacarpals and metatarsals of the 3rd, 4th and 5th digits. Karyotyping revealed 46, X, i (Xq10), while molecular analysis unmasked a silent codon change in exon 5 of GNAS (ATC → ATT, Ile).[8]
In our case, the patient had similar finding on clinical examination except for obesity and heterotrophic subcutaneous calcifications. Biochemical findings were also similar except the patient had microcytic hypochromic anemia. Radiological findings of our case completely correlate with the findings described by Wen Heng-Zeng. Karyotype of our patient showed complete absence of one X chromosome [45,XO].
In our patient, the presence of hypocalcemia, hyperphosphatemia, elevated PTH level along with the normal renal functions and 25(OH) vitamin D levels clearly point towards PHP as the differential diagnosis. Presence of the AHO phenotype points towards the possibility of PHP 1a, 1c and pseudopseudohypoparathyroidism. The presence of AHO along with biochemical abnormalities, presence of renal PTH resistance leaves no other differential diagnosis except PHP 1a and 1c.[1] The Gsα activity analysis further differentiates between PHP 1a and PHP 1c but in the present case, genetic analysis and response to exogenous parathormone could not be done due to financial constraints and lack of available facilities. In the view of clinical history, examination, biochemical, radiological and karyotypic findings, the diagnosis of Turner syndrome with PHP with autoimmune hypothyroidism and microcytic hypochromic anemia was made. The patient was started with calcium, calcitriol, haematinics, thyroxine and estradiol with follow up advice after 2 months. After 2 months of starting therapy, the patient improved clinically and biochemically.
The association of PHP and Turner syndrome is rare. Several heterogeneous and overlapping features like short stature, delayed puberty, hypothyroidism and shortening of metacarpals can lead to a diagnostic dilemma. If unusual features in phenotype like absence of typical facies, shortening of 3rd, 4th and 5th metacarpals and metatarsals etc., then phenotypic features of PHP should be looked for and confirmed biochemically. A thorough clinical history and examination along with judicious use of diagnostic tests can make the diagnosis quite straight forward.
Ethical ConsiderationPatient consent form has been procured prior to the case report study.
Statement of AuthorshipAll authors have given approval to the final version submitted.
Author DisclosureAll the authors have declared no conflict of interest to the work carried out in this paper.
Funding SourceAll expenditures were done by the Department of Endocrinology and Human Metabolism, L.L.R.M. Medical College, Meerut, Uttar Pradesh, India and no external funding organization was involved.
[1] 1. Mantovani G. Pseudohypoparathyroidism: Diagnosis and treatment. J Clin Endocrinol Metab. 2011;96(10):3020-30. DOI.