Medical therapy is indicated in ectopic ACTH syndrome (EAS) when surgery is not feasible, or in cases of severe biochemical disturbances, immunosuppression or mental instability. A cortisol inhibitor such as etomidate may be used in these situations. CASE
We report a rare case of a 41-year-old Malay female with an ectopic ACTH-producing malignant paraganglioma. Our patient presented in February 2014 with an acute stroke. She was also found to be diabetic and hypertensive. Three months later, she was readmitted for severe, symptomatic hypokalemia and poorly controlled diabetes. Her attending physician noted her Cushingoid features. On further inquiry, she had proximal limb weakness, easy bruising, amenorrhea and acne for the past 3 months. She also noticed significant weight loss and insomnia. She did not experience any paroxysms of headache, palpitation and diaphoresis. She was referred to an endocrinologist in a private hospital, who diagnosed her to have ACTH-dependent Cushing’s syndrome. She had markedly elevated midnight serum cortisol [2609 (<50 nmol/L)] and ACTH [62.04 (2.2-13.2 pmol/L)]. Twenty four-hour urine metanephrine level was not elevated. Further imaging using computerized tomography (CT) scan for localization revealed a large lobulated mediastinal mass in the anterior superior mediastinum, mediastinal lymphadenopathy and multiple lung nodules (Figures 1A and 1B). A CT scan-guided fine needle aspiration showed that the mediastinal mass was of neuroendocrine origin, possibly a thymic carcinoid. It stained positive for ACTH on immunohistochemistry. She was then referred to our hospital for further management.
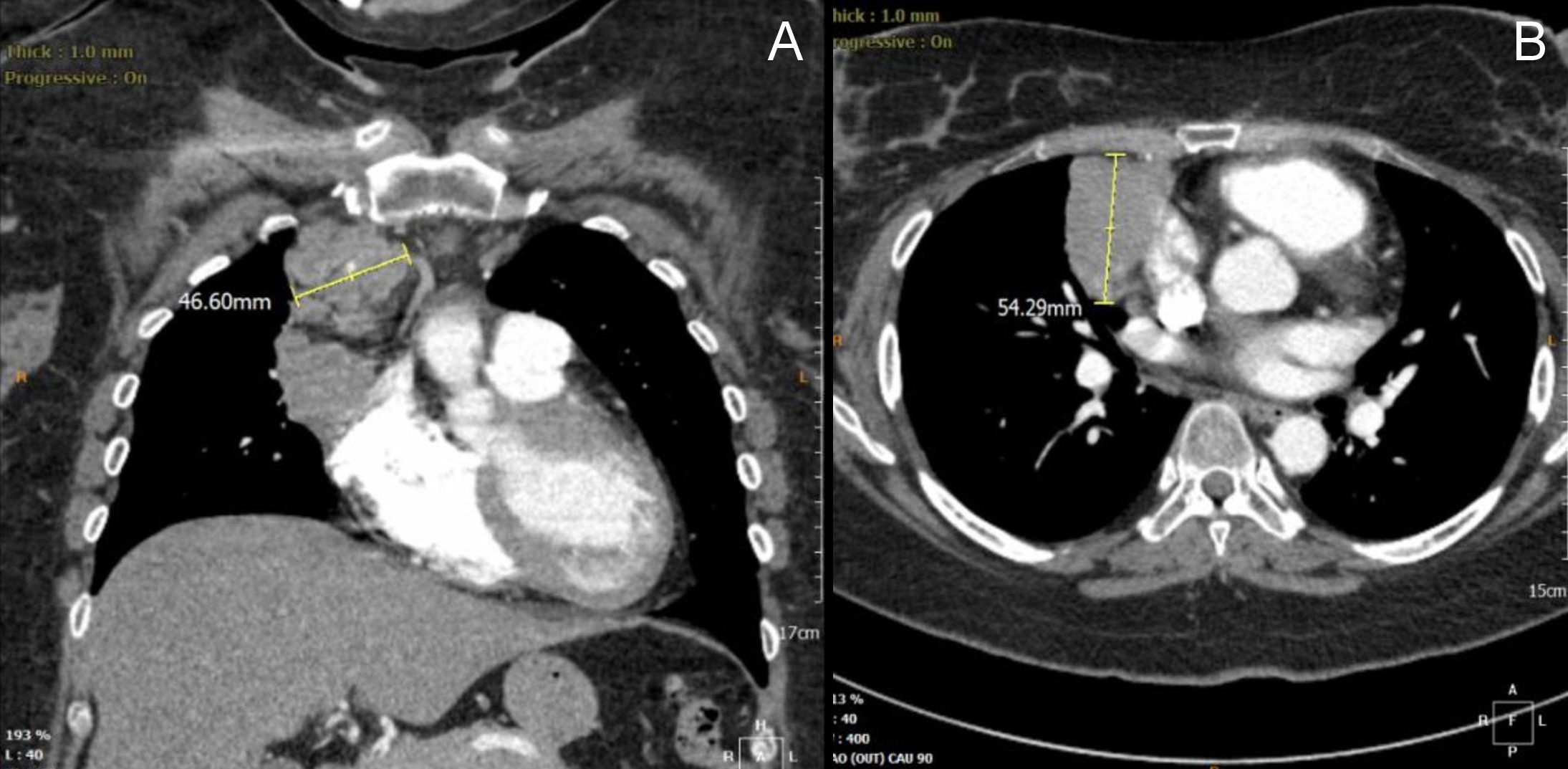
Figure 1. Computerized tomographic scan of the thorax showed a large infiltrative anterior mediastinal mass measuring 5.4 cm x 5.9 cm x 3.8 cm at the anterior superior mediastinum, abutting into right heart margin (coronal view, A). Multiple lung nodules (largest measuring 1.3 cm x 1.1 cm) and mediastinal lymphadenopathy were also noted (axial view, B).
On our assessment, we noted that the patient had truncal obesity, with a body mass index of 29 kg/m2 (weight 70 kg, height 1.55 m). She had prominent hyperpigmentation; most conspicuous over her knuckles, palmar creases and knees; multiple ecchymoses; acne; and mild hirsuitism, with Ferriman-Gallwey score of 9.
Oral ketoconazole was given at 200 mg twice daily and then uptitrated to 400 mg three times a day for control of hypercortisolemia while awaiting definitive therapy. She required continuous potassium replacement, insulin (up to 100 units per day) and 3 anti-hypertensive agents, including spironolactone. While awaiting surgery, she developed a left lung abscess. Blood culture yielded Bacillus sp. This was resolved after bronchial washout and intravenous meropenem and sulfamethoxazole + trimethoprim. She then underwent debulking surgery of her mediastinal tumor on 13 August 2014. Intraoperatively, a large mass infiltrating into the superior mediastinal tissue and over the pericardial surface of right side of heart was seen. It was adherent to right parasternal area and chest wall. Despite the complexities of her surgery, she had an uneventful recovery. Histopathologic examination of the tumor showed malignant mediastinal paraganglioma with heterogenous Ki67 index, ranging from 20% in most areas to 70%. The tumor cells stained strongly positive for neuron-specific enolase, synaptophysin, chromogranin A and CD56.
After debulking surgery, she remained clinically and biochemically Cushingoid despite ketoconazole treatment [ACTH 59.5 (2.2-13.2 pmol/L)]. Metaiodobenzylguanidine (MIBG) scan showed no evidence of MIBG-avid disease. She was then referred to our oncology team. Since the patient declined chemotherapy, she received 30 cycles of external beam radiation which was completed on 15 December 2014. Throughout radiotherapy, her blood glucose control became more challenging, and she had persistent hypokalemia despite regular potassium supplementation. Following radiotherapy, she developed severe nosocomial pneumonia and steroid-related myopathy requiring subsequent invasive ventilation. In view of uncontrolled severe hypercortisolism despite maximal doses of ketoconazole, she was referred for bilateral adrenalectomy.
Intravenous infusion of etomidate was initiated at the recommended low dose of 0.04 mg/hour under close monitoring in the intensive care unit. Vital signs, blood sugar, serum electrolytes and cortisol levels were monitored. We aimed to achieve partial blockade with a target serum cortisol of 500 to 800 nmol/L prior to surgery. Our patient responded rapidly to etomidate: after 8 hours of infusion, cortisol level was halved; after 20 hours, it was relatively low (424 nmol/L), prompting reduction of etomidate infusion rate. Upon the development of hypocortisolism, as indicated by low blood pressure and the need to discontinue intravenous insulin, intravenous hydrocortisone was started. With etomidate infusing at a rate of 1.4 mg/hour (0.02 mg/kg/hour), we managed to achieve a cortisol level of 668 nmol/L just before adrenalectomy (Table 1). The patient underwent bilateral retroperitoneoscopic adrenalectomy on 24 December 2014. Etomidate infusion was stopped prior to induction of anesthesia. As anticipated, blood pressure and sugar control was more manageable after adrenalectomy.

Table 1. Etomidate dosages and parameters monitored
Repeat CT scan on 13 February 2015 showed a slightly smaller residual mediastinal tumor, multiple subcentimeter mediastinal lymph nodes, multiple lung nodules and sclerotic lesions over vertebrae T3, T5, T6, T8, T9 and T11. Our patient unfortunately succumbed one year later in March 2016 after a sudden cardiorespiratory arrest, presumably due to acute pulmonary embolism.
Ectopic ACTH syndrome was first described by Brown in 1928 as “Pluriglandular syndrome: Diabetes of bearded women.”[1] It accounts for 5 to 10% of cases of ACTH-dependent Cushing’s syndrome. EAS is more commonly caused by intrathoracic neoplasms.[2] It typically presents with rapid clinical evolution due to high ACTH levels and the malignant nature of the neoplasm. Apart from the common features of Cushing’s syndrome, anorexia, weight loss and anemia may also be found. Hypokalemia occurs in up to 80% of EAS due to the mineralocorticoid effects of markedly elevated cortisol levels and the decreased activity of 11-hydroxysteroid dehydrogenase type 2.[3] Cushing’s syndrome secondary to ectopic ACTH-producing mediastinal paraganglioma is extremely rare. To date, less than 5 cases have been reported. Mediastinal paragangliomas arise from chromaffin tissue located in the para-aortic ganglia, with a tendency to invade bordering structures as observed in our patient. Fifty percent of patients are asymptomatic and incidentally diagnosed.[4] Other manifestations include mass effects and hormonal hypersecretion.
Surgical clearance of tumor is the only curative measure in EAS. In cases where surgery is not feasible, medical therapy to control hypercortisolemia is imperative. Other indications of medical therapy include severe biochemical disturbances, such as hypokalemia; immunosuppression; mental instability; or following radiotherapy. Bilateral adrenalectomy may be considered in patients with severe hypercortisolemia or intolerance to oral therapy.[5] Surgical risks may be significantly reduced if cortisol levels are normalized preoperatively. Etomidate is a carboxylated imidazole which inhibits mitochondrial cytochrome P450-dependent enzyme 11β-hydroxylase that catalyzes cortisol conversion from deoxycortisol. It was initially developed as an intravenous hypnotic non-barbiturate induction anesthetic agent, but was noted to increase mortality in critically unwell patients and cause low serum cortisol. The starting dose is 0.04 to 0.05 mg/kg/hour (2.5 to 3.0 mg/hour). Etomidate initiation should be monitored in the ICU setting for close monitoring of plasma cortisol and potassium. Intravenous hydrocortisone may also be used in a “block and replace” strategy, with a serum cortisol target of 500 to 800 nmol/L.[6] There is a clear delineation between higher anesthetic dose and lower doses which inhibits adrenal function. Schulte showed that etomidate only causes prominent sedation at the highest dose of 0.3 mg/kg/hour. In their protocol, etomidate is started at 2.5 mg/hour regardless of body weight, and titrated up to 4 mg/hour according to cortisol level.[7] Etomidate is an effective treatment for hypercortisolism, but is limited to short-term use. It is generally used to “buy time” while awaiting other definitive therapy.
Our patient required a much lower dose of etomidate compared to other protocols. As with many other treatments, the dosages required by Asian patients tend to differ from their Caucasian counterparts. This is possibly due to ethnic differences in the metabolism of medications. From our own experience, we will continue to use a lower starting dose of etomidate on our patients in the future. Close clinical and biochemical monitoring of patients to enable appropriate dose adjustment is essential.
Low dose etomidate can be effectively used to control severe hypercortisolism.
Ethical ConsiderationInformed consent has been taken before submission of the manuscript.
Statement of AuthorshipAll authors certified fulfillment of ICMJE authorship criteria.
Author DisclosureThe authors have declared no conflict of interest.
Funding SourceNone.
[1] Brown WH. A case of pluriglandular syndrome: Diabetes of bearded women. Lancet. 1928;2:1022-3.