Blepharophimosis ptosis epicanthus inversus syndrome (BPES) is a rare congenital eyelid disorder that is inherited as an autosomal dominant trait, with an estimated incidence of 1 in 50,000 births.[1] Blepharophimosis was first reported by Von Ammon in 1841. Vignes first associated blepharophimosis with ptosis and epicanthus inversus in 1889. It is characterized by shortened horizontal palpebral fissure (blepharophimosis), impaired function of levator palpebrae superioris of upper eyelid (ptosis), a vertical skin fold arising from the lower eyelid that inserts medially into the upper lid (epicanthus inversus) and an increased inner canthal distance (telecanthus).[2] Zlotogra et al. in 1983 described two types of BPES, Type I and Type II. Type I is associated with premature ovarian failure (POF) in the affected female, in addition to the classical eye findings, while Type II has only eye features.[3]
The diagnosis of BPES is primarily made by combination of typical facio-ocular features, with clinical and biochemical features of primary ovarian insufficiency. For diagnostic purpose, genetic analysis is not needed. However, both types of BPES are caused by mutations of the forkhead transcriptional factor 2 (FOXL2) gene, that is located on the long arm of chromosome 3 (3q23).[4]
In this paper, we report a family of two female siblings, who have all the eye manifestations of BPES along with amenorrhea, while their father has only ophthalmic manifestations.
We describe an Asian Indian family of 2 female and 2 male siblings. Both sisters, who were 22 years and 19 years of age, presented to the Endocrinology OPD, with complaints of bilateral drooping of the eyelids, dimness of vision since early childhood and secondary amenorrhea for 4 years and 2 years, respectively.
There was no history of puffiness of face, swelling of eyes, constipation, deafness, proximal myopathy, any chronic illness, mental retardation, chemotherapy, radiation, pelvis surgery, absence of tears and hyperpigmentation. Their antenatal, natal and postnatal histories were unremarkable. General examination revealed normal stature with absence of low set ears, short fourth metacarpal, shield chest, cubitus valgus, web neck and low posterior hairline. Both sisters have normal secondary sexual characteristics (Tanner stage A+ P4 B5) and attained menarche at age of 14 years. On ophthalmologic examination there was bilateral ptosis, blepharophimosis and epicanthus inversus (vertical skin fold arising from the lower eyelid that inserted medially into the upper lid). The length of both horizontal palpebral fissures were 19 mm in the older sister and 20 mm in the younger sister, which were less than the normal adult Indian female measurement of 33.7±1.8 mm. The length of both vertical palpebral fissures were 4 mm in the older sister and 3.5 mm in the younger sister, which were less than the normal adult Indian female measurement of 11.7 ± 1.6 mm. The distance between the medial canthi (telecanthus) was 38 mm in the older sister and 39 mm in the younger sister, which were more than the normal adult Indian female measurement of 32.7 ± 1.5 mm (Table 1) (Figure I).[5] Visual acuity was decreased in both eyes. The father also has similar eye findings of blepharophimosis, ptosis, epicanthus inversus and telecanthus (Figure 2).
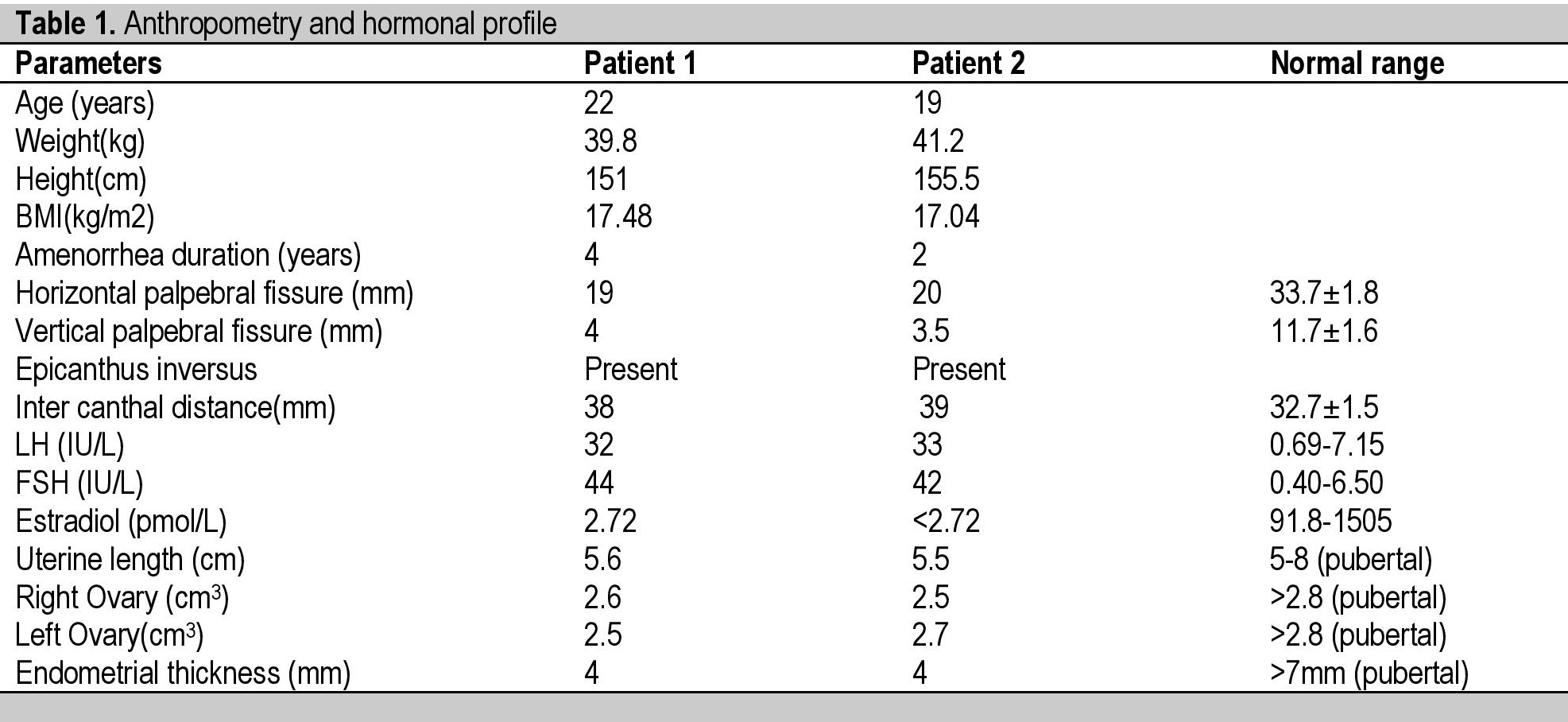
Table 1. Anthropometry and hormonal profile
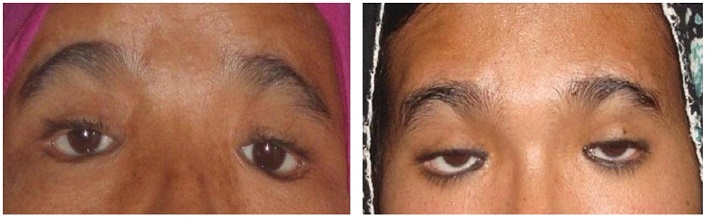
Figure 1. Characteristic clinical features of the patients with blepharophimosis, ptosis, epicanthus inversus syndrome (BPES). Both patients have small palpebral fissures, ptosis of the eyelids, and epicanthus inversus.
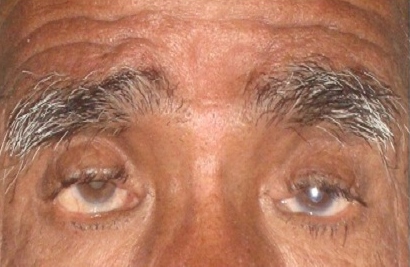
Figure 2. Characteristic clinical features of the patients with blepharophimosis, ptosis, epicanthus inversus syndrome (BPES). The father also has small palpebral fissures, ptosis of the eyelids, and epicanthus inversus.
Laboratory investigations revealed normal hemogram, renal function test, hepatic function test, electrolytes, and prolactin. Investigation also revealed raised FSH values above 40 IU/L on two occasions, 1 month apart, with raised LH and low estrogen. Ultrasound of the pelvis showed atrophic ovaries, small uterus with endometrial thickness of 4 mm in both sisters. Chromosomal analysis revealed normal 46,XX karyotype. Cytogenetics and molecular genetic analysis were not done due to unavailability and financial constraints. All the hormonal investigations were done by chemiluminescence immunoassay using Abbott ARCHITECT i1000sr immunoassay analyzer.
On the basis of typical clinical history, examination findings and laboratory evaluation, a diagnosis of type 1 BPES was formed in both sisters and they were started on hormone replacement therapy (estrogen and progesterone) with calcium and vitamin D supplementation. Need for eye surgery and fertility issue were discussed. Written informed consent from to publish the case report and images has been taken and is approved by the local ethics committee.
BPES is a complex eyelid malformation, characterized by four major features: blepharophimosis (narrowing of horizontal aperture of the eyelids), ptosis (drooping of upper eyelid), epicanthus inversus (a skin fold arising from the lower eyelid and running inwards and upwards) and telecanthus (lateral displacement of the inner canthi with normal interpupillary distance). BPES type I includes the four major features and POF; BPES type II includes only the four major features. Other ophthalmic manifestations that can be associated with BPES include lacrimal duct anomalies, amblyopia, strabismus, and refractive errors. Minor features include a broad nasal bridge, low set ears, and a short philtrum.[4],[6]
The definition of POF is amenorrhea, hypoestrogenism, and elevated serum gonadotropins in women less than 40 years of age. More than 4 months of amenorrhea and two serum FSH levels of more than 40 IU/L obtained more than one month apart in women aged <40 years are the suggested criteria for diagnosing POF.[7]
In the Indian population, mean age of BPES cases was 12 ± 8.4 yrs (range 4-32 years), with the majority of cases between 4-8 yrs. This finding is consistent with studies from other parts of the world that have reported that majority of cases present before 8 yrs of age. In our case, patients present at 22 and 19 yrs of age after the onset of amenorrhea.[1]
Townes and Muechler, Fraser et al., Smith et al., and Panidi et al., described families affected with POF and eye features of BPES.[8],[9] ,[10]
The genetic pathophysiology of BPES is due to a FOXL2 gene mutation which is responsible for BPES type I and II, which is located on the long arm of chromosome 3 (3q23). Four types of deletions in chromosome 3q has been described in BPES (46,XY,del 3qter; 46,XY,del 3q26.3; 46,XX,del 3q24-25 and 46,XY,del 3q26-qter). Patients who are cytogenetically normal are further evaluated for molecular analysis for FOXL2 sequence variations. Complete or partial loss of FOXL2 protein function leads to development of BPES types I and II respectively. The FOXL2 gene instructs the proteins involved in eyelid muscles and ovarian development. More than 100 FOXL2 gene mutations have been identified in BPES, which includes frameshift insertions, nonsense mutations and missense mutations.[1]
Mati et al., showed that the form of BPES associated with premature ovarian failure maps to 3q22-q23, the same chromosomal region as does the form without POF.[11] By positional cloning, Crisponi et al., identified the FOXL2 gene and identified mutation resulting in truncated proteins in affected individuals with both Type I and II BPES. FOXL2 was selectively expressed in the mesenchyme of developing mouse eyelids and in adult ovarian follicles; in adult humans, it appeared predominantly in the ovary.[12] In our patients, cytogenetic and molecular analysis could not be done beyond karyotyping because of financial constraints.
Treatment of BPES is eyelid surgery which involves a medial canthoplasty for correction of the blepharophimosis, epicanthus inversus, and telecanthus at age three to five years, typically followed a year later by ptosis correction, a one-stage procedure has also been described. Premature ovarian failure is treated with hormone replacement therapy (estrogen and progesterone), fertility is addressed with reproductive technologies such as embryo donation and egg donation.[6]
BPES is usually inherited in an autosomal dominant manner but autosomal recessive inheritance has also been reported in one consanguineous family. Prenatal testing for pregnancies at increased risk is possible if the pathogenic variant in the family has been identified.BPES is usually inherited in an autosomal dominant manner but autosomal recessive inheritance has also been reported in one consanguineous family. Prenatal testing for pregnancies at increased risk is possible if the pathogenic variant in the family has been identified.[6]
BPES can either present as only typical eye findings or combined with primary ovarian failure. Therefore, the possibility of BPES must be borne in mind in any patient presenting with POF and associated characteristic eye features. If available, molecular characterization and genetic evaluation may be useful for a more definitive diagnosis and genetic counseling. Family pedigree construction is also important to identify the pattern of inheritance.
Ethical ConsiderationInformed consent has been taken before submission of the manuscript.
Statement of AuthorshipAll authors certified fulfillment of the ICMJE authorship criteria.
Author DisclosureThe authors have declared no conflict of interest.
Funding SourceNone.
[1] Chawla B, Bhadange Y, Dada R, Kumar M, Sharma S, et al. Clinical, radiologic, and genetic features in blepharophimosis, ptosis, and epicanthus inversus syndrome in the Indian population. Invest Ophthalmol Vis Sci. 2013;54(4):2985-91. PubMed DOI.