Congenital adrenal hyperplasia is a leading cause of 46,XX disorders of sex development (DSD) resulting from a deficiency of enzymes required for cortisol and aldosterone biosynthesis. More than 90% cases have 21-hydroxylase deficiency (21-OHD) and mutations in the CYP21A2 gene, which is located on the short arm of chromosome 6 (6p21.3).[1],[2] Affected females are born with various degrees of virilization, as a result of prenatal androgen exposure. Aside from 21-OHD, virilization in 46,XX individuals with CAH is also caused by 11β-hydroxylase deficiency (11β-OHD), P450 oxidoreductase deficiency (POR) and 3β-hydroxysteroid dehydrogenase deficiency (3β-HSD).[2],[3],[4]
Classic CAH can be further divided into salt-wasting and simple virilizing types. Virilization in a female manifests as clitoromegaly, acne, hirsutism and low voice. At puberty, normal feminization of girls fails to occur, usually presenting with lack of breast development or absence of menstruation.[3],[4] Elevation of 17-OHP in CAH leads to increased androgen levels, but the relationship between 17-OHP and virilization phenotype has not been consistent as seen in previous studies.[2],[5]
The incidence of CAH in the general population is approximately 1 in 16,000 on screened newborns worldwide, and carriers are present in 1:60.[6],[7] Data from our center from 2004 to 2016 show that there are 84 patients with CAH, consisting of a wide age range of 3 days to 44 years old. Majority of the patients seen have never received any specific treatment for CAH. Genital ambiguity was the most common reason for physician consult in female patients.
Delayed diagnosis and treatment of CAH in children is common in Indonesia. It is frequent especially for SV or SW types who develop adrenal crises without a definite diagnosis due to the absence of newborn screening.[8] In Indonesia, newborn screening for CAH is not yet available, as basic treatment and hormonal tests are not provided in public hospitals with health insurance services. Ethically, it cannot be provided yet because medications such as hydrocortisone and fludrocortisone are not available. In developed countries, a patient with CAH is often diagnosed early in life by neonatal screening programs.[9]
OBJECTIVES
1. To describe the phenotype variation in Indonesian 46,XX late-identified CAH, which is rarely reported in other countries, and;
2. To identify the correlation of 17-OHP and genital virilization.
METHODOLOGYStudy Design
This is a descriptive study.
Patients and Methods
From 2004 to 2016, we gathered 84 patients with a diagnosis of CAH, evaluated under the clinical management of the multidisciplinary DSD team of the Dr. Kariadi Hospital and Faculty of Medicine Diponegoro University. Thirty-nine patients were enrolled in this study.
CAH was diagnosed based on the clinical manifestations and elevated serum levels of 17-OHP. The inclusion criteria were 46,XX karyotype and age 6 months or older at first visit to our center. Patients who underwent corrective genital surgery before their first visit to our center were excluded.
Medical records were reviewed to gather data including age at diagnosis, anthropometrics, appearance of genitalia, Prader score, Tanner stage, skin hyperpigmentation, acne, hirsutism, and low voice with or without Adam’s apple, menstrual disorders and family history (pedigree). Details and additional information were verified with the patient or parent.
Ethical Considerations
This study was approved by the Medical Ethics Committee and informed consent was obtained from all patients and parents.
Clinical Findings
Thirty-nine patients (5 SW and 34 SV) fulfilled the inclusion criteria, with a mean age of 9.83 (SD±9.42 years). There were 9 adults and 30 children. Physician-diagnosed SW type was significantly younger compared to SV type (mean age 0.58 versus 7.25 years, p<0.05). The phenotype variation of late identified CAH are shown in Table 1. The most frequent clinical manifestations on both SW and SV types were clitoromegaly (100%) and generalized skin hyperpigmentation mainly around the genital area (87%). Nine patients with ages over 13 years showed lack of breast development (breast Tanner stage 1 to 2). Menstrual disorders included primary amenorrhea (67%) and late menarche occurring at age over 15 years (33%, 15 to 18 years). Precocious puberty (23.3%), defined as premature pubic hair growth, was evident by 3 years old. Fourteen patients had low voice (35.9%), 9 of which had prominent Adam’s apple. Four patients had hirsutism (10.3%), all of whom were SV type. Two patients had acne problems. Eight children had height measurements above the 97th percentiles for age in the Chinese Growth chart (26.7%), the available reference for Asian populations, indicative of rapid growth.[10] Short stature, defined as less than the 3rd percentiles of the Chinese Growth Chart, occurred in 6 adult patients (75%). Of this group, one child had accelerated epiphyseal maturation, with bone age at 17 to 18 years.

Table 1. Phenotypic variation of Indonesian late-identified CAHa patients
The next most common dysmorphology was rudimentary labia minora, seen in 80% of SW and 67% of SV types (p>0.05). However, clitoral/phallus length was not significantly different between SW and SV types, with mean length 4.8 cm and 4.24 cm, respectively (p>0.05). We found a good correlation between age and phallus length in late-identified CAH (r=0.466, p<0.05) (Figure 1). More than 50% of both SW type and SV type CAH had labial scrotalization. Complete labial fusion was more frequent in SW (20%) compared to SV (17.9%) (p>0.05).
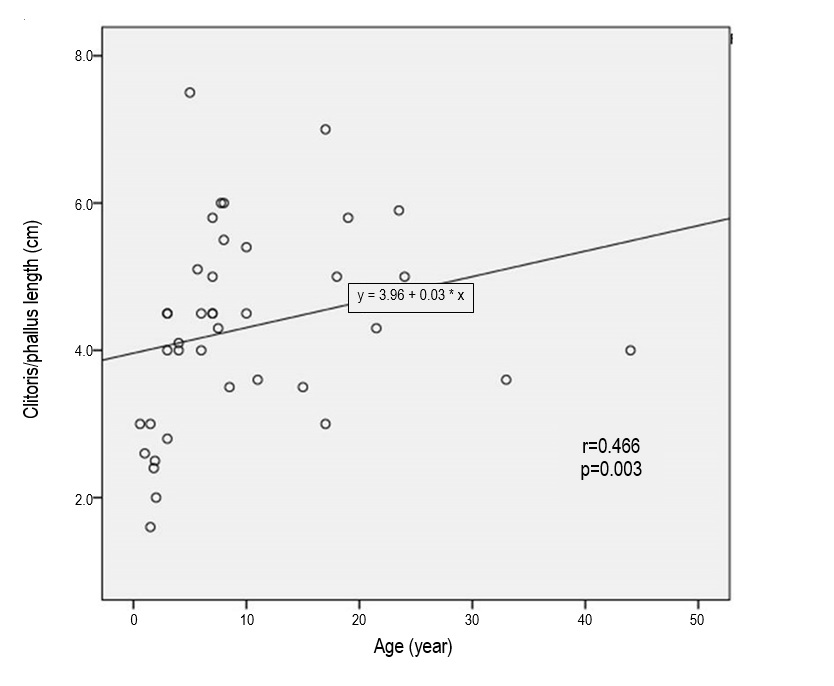
Figure 1. Relationship between age and phallus length in late-identified CAH.
All patients had genital ambiguity, with a median Prader score of 3 and 4 in SW and SV types, respectively. Thirteen patients (33%) reveal male gender and four patients with male gender of five SW-type on their first visit and one SV type patient had undetermined gender. However, after undergoing thorough physical and laboratory examination and genetic counseling, four patients decided to change their gender from male to female (Table 2).

Table 2. Gender assignment after final examination
Correlation of 17-OHP with Phallus Length
All patients had elevated blood levels of 17-OHP. Based on Spearman’s correlation coefficient test, there was no correlation between 17-OHP level and phallus length (r=0.19, p>0.05) (Figure 2).
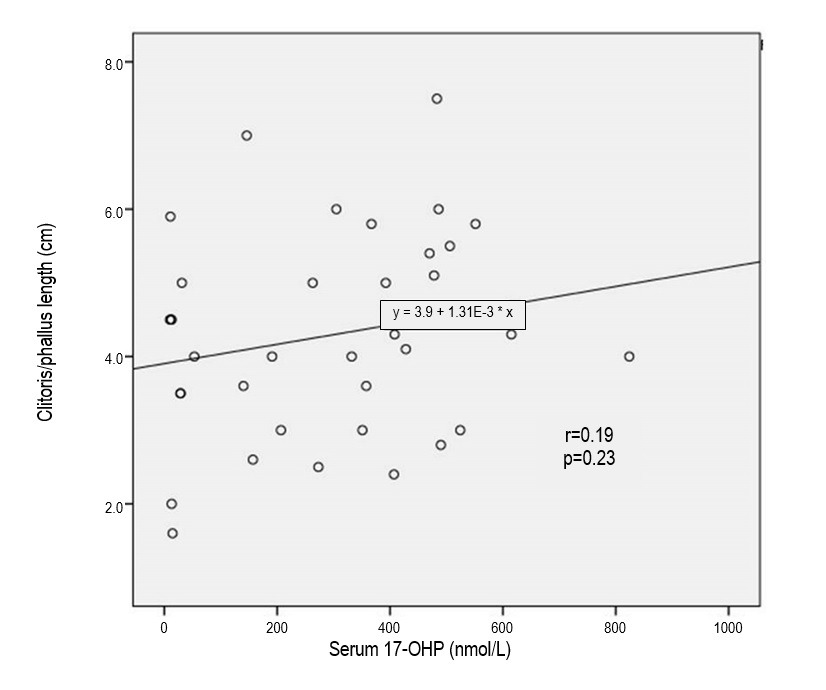
Figure 2. Relationship between serum 17-hydroxyprogesterone (17-OHP) levels and phallus length in late-identified CAH.
The mean age of presentation of patients in our study is nine years, quite different from data from Western populations.[11] Recognition is frequently late due to lack of knowledge of medical providers, especially in remote regions; lack of awareness of parents to seek medical management; unavailability of neonatal screening in Indonesia; and early death of affected babies before diagnosis.
There are significant differences in the number of patients and their age at diagnosis in SW and SV types in our study, in contrast to other published reports.[12] This discrepancy suggests that more of SW type CAH patients died from adrenal crises before being diagnosed. Identifying newborn girls with signs and symptoms of CAH is not difficult because of genital ambiguity and salt wasting symptoms in the SW type. In newborn boys, the diagnosis is challenging because of the absence of ambiguous genitalia, especially in the SV type. Moreover, CAH in Indonesia is still unfamiliar for many medical practitioners, such as general practitioners or midwives. Factors that may lead to late diagnosis and treatment include high cost and limited facilities for hormonal analysis to detect CAH, and the difficult access to appropriate medications.
Clinical variations depend on the degree of hormonal disruption, and timeliness and regularity of treatment.[13] Virilization is very prominent in late-identified females with CAH due to the untreated condition (Figure 3). There is a good correlation between age and phallus length in late-identified CAH, indicating that older untreated CAH females are more virilized. Androgen exposure in these girls start from the prenatal period and persist until they receive treatment.[14] In our study, androgen excess in untreated females resulted to progressive virilization of the clitoris, hyperpigmentation, lack of breast development, primary amenorrhea, delayed menarche, premature pubic hair growth, hirsutism, low voice with or without prominent Adam’s apple and short stature. These observations are comparable with findings in previous studies.[15],[16]
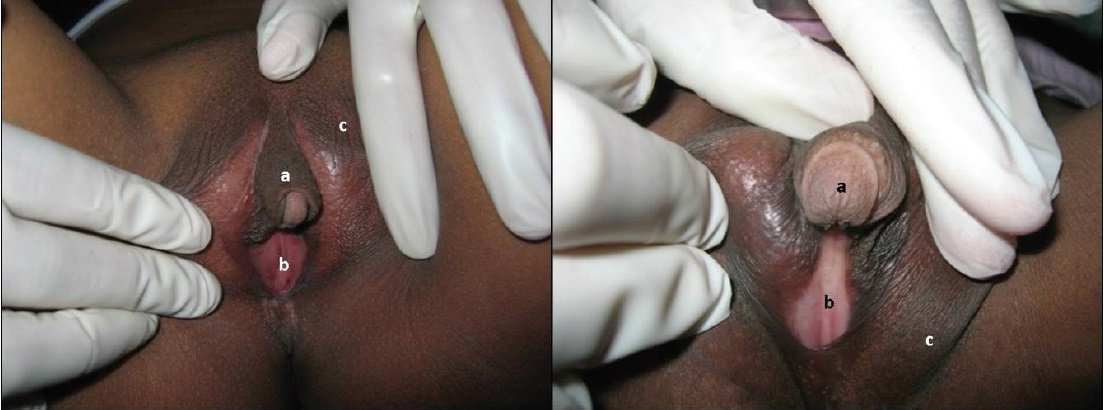
Figure 3. Virilized external genitalia in 2 female siblings showing (a) clitoromegaly, (b) presence of vaginal introitus (c), and hyperpigmentation.
In our study, thirty-three percent of patients were considered male because of the presence of male-looking genitalia (clitoromegaly resembling penis, scrotalization with or without complete labial fusion) and the absence of treatment (Figures 4 and 5). This emphasizes the need for a thorough urogenital examination in the neonatal period for patients with DSD. Chromosomal and hormonal analyses should be performed on neonates with ambiguous genitalia to facilitate gender assignment.[17]
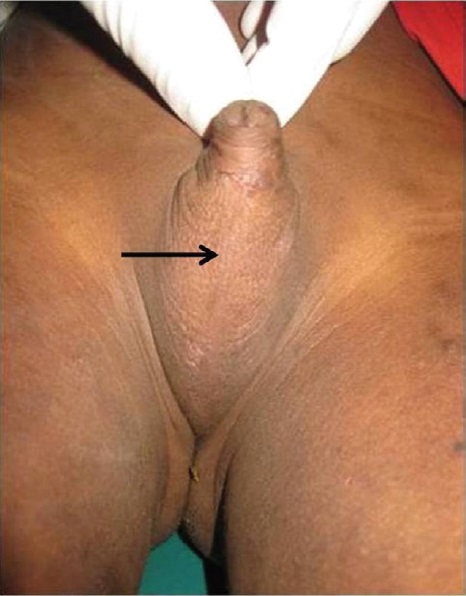
Figure 4. An 8.5-year-old child assigned to male gender with 46,XX late-identified CAH. There is complete labial fusion (arrow) resembling a scrotum.
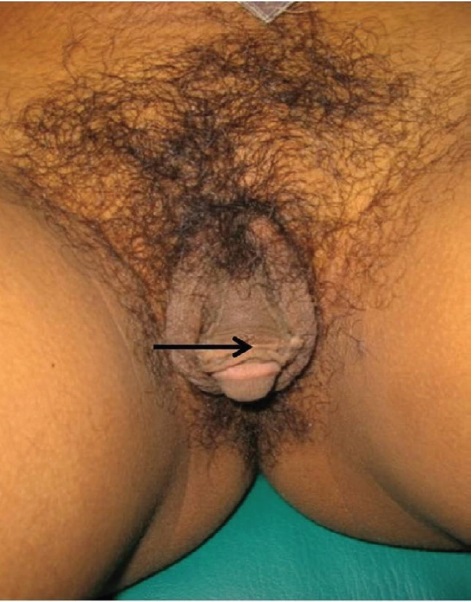
Figure 5. A 24-year-old adult assigned to male gender with 46,XX late-identified CAH. There is a big phallus (arrow) with scrotalization.
Precocious puberty in female CAH patients are recognized by premature pubic hair growth occurring before 8 years of age and rapid growth.[18] In the same study, patients treated after age one year have a higher risk for precocious puberty; for SV type boys, this risk was apparent even in those who received treatment at the age of 6 months.[18] We found similar findings with Hargitai et al., our SV type children showed accelerated growth patterns after 3 years old, while the final height of adult patients leading to short stature was reduced compared to the standard population and their respective target heights.[19] Bone age determination may be able to show accelerated epiphyseal maturation. Unfortunately, these results were not available in all our CAH children, due to financial constraints and limited facilities.
In our study, all late-identified CAH patients had elevated blood levels of 17-OHP. The 17-OHP levels were weakly correlated with phallus length (r=0.19). However, there was no clear relationship between 17-OHP levels and virilization of external genitalia, similar to findings by Rocha et al.[20]
Genetic counseling for families and patients is advised to explain the clinical manifestations, mode of inheritance, recurrence risk and consequences of CAH. Some challenges to our work include varied cultural beliefs within families, patient discrimination and taboos pertaining to their masculinized condition, and the unavailability of a genetic counselor.[21] Gender assignment is still a dilemma: some of the children were raised as males, females or left undecided. Late-identified CAH females with progressive virilization should undergo psychological evaluation to assess their emotional condition and gender identity.
In our study, the most common clinical features in late-identified Indonesian CAH patients were clitoromegaly and skin hyperpigmentation. Clinical features such as genital virilization, low voice, Adam's apple and short stature in adults were permanent phenotypes, indicating the severity and irreversibility of virilization despite medication. There was no correlation between of 17-OHP levels and virilization. There is a general lack of awareness of medical personnel, family members and patients regarding CAH and its effects. The high cost and limited availability of laboratory tests and difficult access to medications pose significant challenges as well. Improvement in the clinical recognition of suspected CAH patients is important in our Indonesian clinical setting. Genetic counseling is crucial in the management of CAH, especially for gender determination. Newborn screening should be included in the improvement of health policy in Indonesia.
AcknowledgmentsWe thank the multidisciplinary DSD team of Dr. Kariadi Hospital - FMDU and the staff in CEBIOR for their help with the patients and laboratory work. We would like to thank all the families involved in our study. The author (MU) is a recipient of the Beasiswa Unggulan BPKLN fellowship from the Indonesian Ministry of Education and Culture.
Statement of AuthorshipAll authors certified fulfillment of ICMJE authorship criteria.
Author DisclosureThe authors declared no conflict of interest.
Funding SourceThis study was supported by Competitive Research Grant from the Indonesian Ministry of Research, Technology and Education - DIPA number 023.04.2.673453/2015 and number 022/SP2H/LT/DRPM/II/2016.
[1] Ekenze SO, Nwangwu EI, Amah CC, Agugua-Obianyo NE, Onuh AC, Ajuzieogu OV. Disorders of sex development in a developing country: Perspectives and outcome of surgical management of 39 cases. PediatrSurg Int. 2015;31(1):93-9. PubMed CrossRef.