Adrenocortical carcinoma is a rare malignancy with an incidence of approximately one to two per million population per year.[1] Sixty two percent of adrenocortical carcinomas present as functioning tumors with the most common presentation being Cushing’s syndrome or a combination of hypercortisolism and virilization.[2] Aldosterone-producing adrenocortical carcinoma is even more uncommon, comprising only <7% of all functioning adrenocortical carcinomas.[3] A complete hormonal evaluation is needed prior to surgery and is necessary to establish tumor markers for monitoring of tumor recurrence. Management involves complete surgical resection of the tumor followed by chemotherapy depending on the clinical staging. Lifelong monitoring for tumor recurrence is recommended. This report presents a rare case of aldosterone-producing adrenocortical carcinoma with cosecretion of cortisol and estradiol with a clinical picture of primary aldosteronism and subclinical Cushing’s syndrome and feminization. A review of literature related to the diagnosis and management of adrenocortical carcinoma is also discussed.
CASEAn 18-year-old male presented with intermittent bilateral leg weakness and uncontrolled hypertension. He is a known hypertensive maintained on 5 antihypertensive medications. History started 3 years prior to admission when the patient was noted to have BP elevations, highest was at 140/90 mm Hg, with no associated symptoms. Consultation with a pediatrician was done and he was prescribed with Amlodipine 5 mg OD and Metoprolol 50 mg OD which he only took for less than a month. No work-up was performed. Two years prior to admission, after undergoing high intensity volleyball training for 3 straight hours, the patient experienced weakness of both lower extremities which progressed to inability to move his lower extremities after a day. He consulted after 5 days and was admitted for hypokalemia (serum potassium 2.7 mmol/L) and hypertension (BP 180/100 mm Hg). Serum potassium was corrected and he was discharged improved with the following medications: Amlodipine 10 mg OD and Metoprolol 50 mg OD. The patient was again not compliant with his medications and continued to experience intermittent body weakness until, 1 year prior to admission, the patient experienced loose bowel movement and was admitted and diagnosed to have amoebiasis. During this admission, the patient was again noted to have hypokalemia (serum potassium 2.7 mmol/L) and hypertension. Potassium correction and BP control were done. Further work-up showed mirror image dextrocardia and left atrial and ventricular enlargement on 2D echocardiogram and situs inversus with normal kidneys on ultrasound. The patient was discharged improved with the following medications: Losartan 50 mg OD and Potassium Chloride tablet TID and was told to follow up with a cardiologist. On follow-up, the patient was still noted to have BP elevations hence the following medications were subsequently added: Spironolactone 100 mg OD, Verapamil 180 mg OD, Clonidine 150 mcg OD, and Terazosin 5 mg OD. He was then referred to an endocrinologist for further work-up.
He has no headache, increased sweating, palpitations, weight changes, easy bruisability, moon facies, excessively oily nor acne-prone skin, hair loss over the axillary and pubic areas, decreased hair growth, nipple discharge, breast tenderness, decreased libido, or impotence. He is obese with gynecomastia and minimally scattered violaceous striae over the abdomen.
Given a young patient with uncontrolled hypertension on more than 3 anti-hypertensive medications, intermittent bilateral leg weakness and history of hypokalemia, the initial impression then was hypertension most probably due to primary aldosteronism. Thyroid function tests, creatinine, blood sugar, lipid profile, complete blood count, and ultrasound of the kidneys were all normal. A 2D echocardiogram showed mirror-image dextrocardia, left atrial and ventricular enlargement with normal ejection fraction. Laboratory tests showed an elevated plasma aldosterone concentration (PAC) of 47.66 ng/dL (normal value 4.20-20.15), suppressed plasma renin activity (PRA) of 0.04 ng/ml/hr (normal value 0.30-1.90), elevated aldosterone/renin ratio (ARR) (1,191.5), elevated 24 hour urine metanephrine (6.8 mg/24 hours, normal <1.0) and normal plasma free metanephrine (5.650 pg/ml, normal <90.00) (Table 1). Since the patient had undetectable renin, PAC >20 ng/dL and spontaneous hypokalemia, there was no need for confirmatory testing.[4] CT scan with adrenal protocol showed a well-defined, enhancing hypodense focus measuring 4.7 x 4.1 x 4.8 cm in the left suprarenal region, indenting on segment 6 of the right liver lobe (Figure 1). It has an average HU of 26 in the unenhanced phase, 52 HU in the adrenal/venous phase, and 35 HU in the delayed phase with an absolute washout of 65.4% suggestive of a lipid-poor tumor. The 24-hour urine free cortisol was elevated at 131.20 ug/24 hours (normal value: 20.00-90.00), nonsupressible cortisol after 1 mg dexamethasone (2.8 ug/dL, normal value <1.8), normal basal cortisol (10.00 ug/dL), decreased basal ACTH (0.25 pg/ml, normal value 10-90), elevated estradiol (60.00 pg/ml, normal value 11-44) and normal DHEA-S (176.10 ug/dL, normal value 45.10-385). Since the patient was young (<age 35) with spontaneous hypokalemia, marked aldosterone excess, and unilateral adrenal mass, adrenal venous sampling was not performed before proceeding with adrenalectomy.[4]
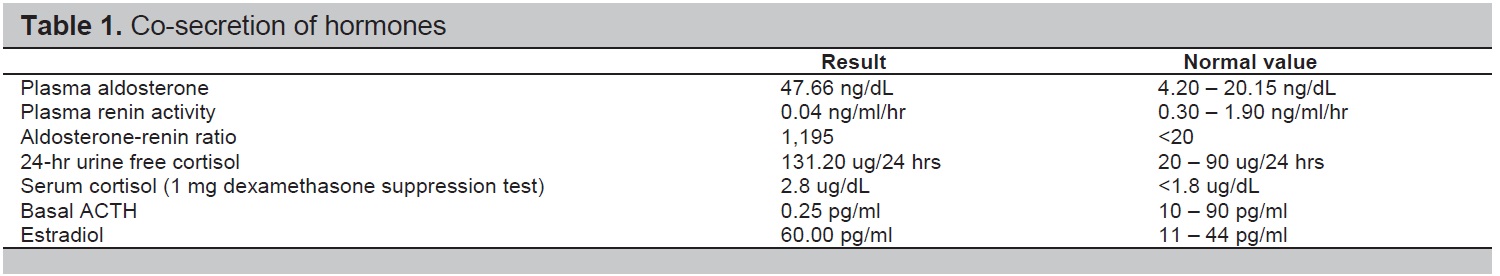
Table 1. Co-secretion of hormones
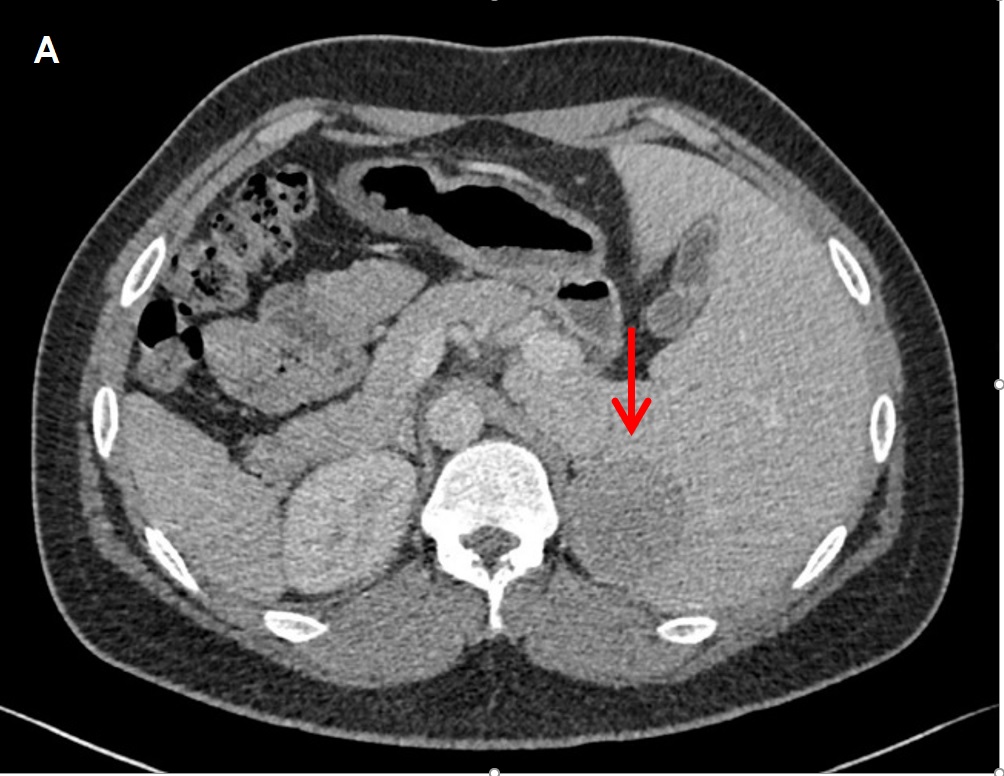
Figure 1. Computed tomography scans taken during the adrenal/venous phase (60 seconds after contrast injection). A well-defined, enhancing hypodense focus measuring 4.7 x 4.1 x 4.8 cm is seen in the left suprarenal region. (A) Axial view
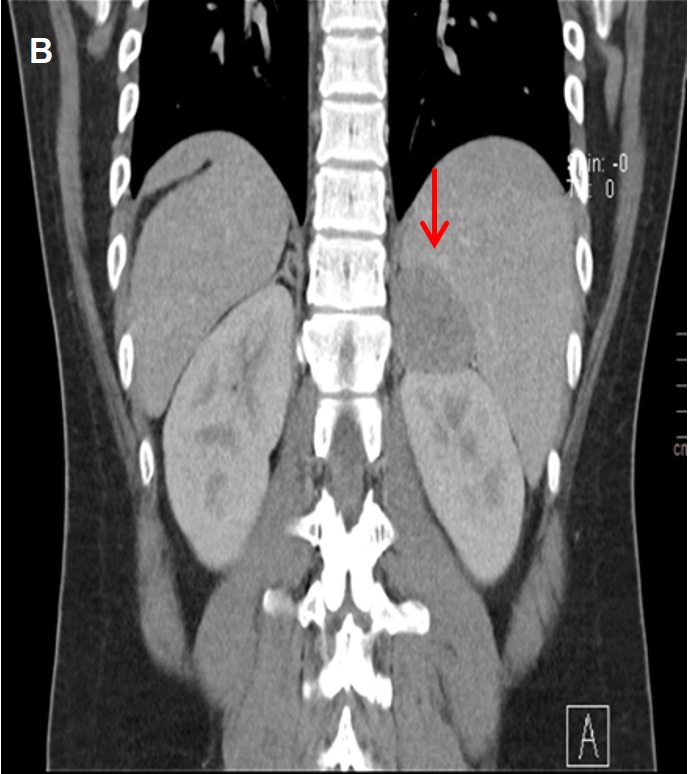
Figure 1. Computed tomography scans taken during the adrenal/venous phase (60 seconds after contrast injection). A well-defined, enhancing hypodense focus measuring 4.7 x 4.1 x 4.8 cm is seen in the left suprarenal region.(B) Coronal view.
Laparoscopic adrenalectomy was done with steroid coverage. The specimen weighed 62 grams and measured 7.0 x 6.4 x 4.3 cm (Figure 2A). Serial sections of the specimen showed a well-defined mass measuring 6.0 cm in its widest diameter. It has pink-tan to yellow soft cut surface with hemorrhagic areas (Figure 2B). Histopathologic diagnosis was malignant adrenal neoplasm (adrenocortical carcinoma versus pheochromocytoma) based on the Modified Weiss criteria.
Immunohistomorphology supported the diagnosis of adrenocortical caricnoma. The Ki67 index, a marker of proliferative activity, was 5-10%. No further treatment such as chemotherapy was done after complete surgical resection. His blood pressure decreased and aldosterone, cortisol and estradiol levels returned to normal. He was told to follow up regularly for monitoring of tumor recurrence.
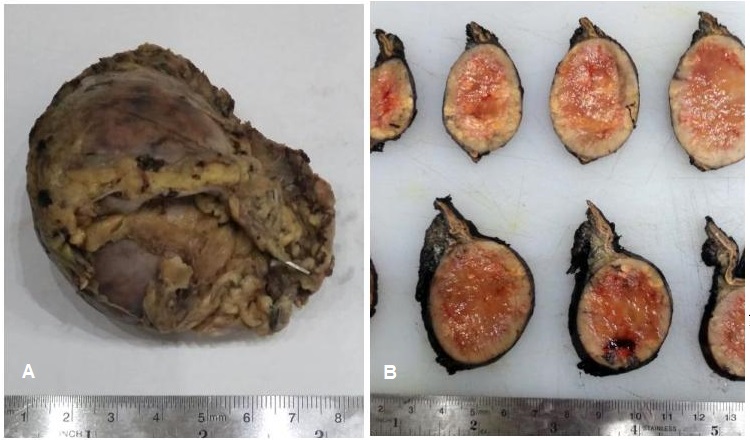
Figure 2. Gross appearance of the left adrenalectomy specimen. (A) The specimen weighed 62 grams and measured 7.0 x 6.4 x 4.3 cm, brown tan to gray and partially covered with fibrofatty tags. (B) Serial sections of the specimen showed a well-defined mass measuring 6.0 cm in its widest diameter with pink-tan to yellow soft cut surface and hemorrhagic areas.
Adrenocortical carcinoma is a rare malignancy with an incidence of 1 to 2 per million population per year.[1] The most common presentation of adrenocortical carcinoma is Cushing’s syndrome or a combination of hypercortisolism and virilization. Only less than 40% would present as nonfunctioning tumors.[3] Aldosterone-producing adrenocortical carcinoma is even more uncommon presenting as uncontrolled hypertension and severe hypokalemia. The patient initially presented with uncontrolled hypertension and intermittent bilateral leg weakness due to hypokalemia. A high plasma aldosterone concentration and suppressed plasma renin activity confirmed the diagnosis of primary aldosteronism. Aldosterone-producing adenomas and bilateral idiopathic hyperplasia are the most common causes of primary aldosteronism. Only less than 1% are due to aldosterone-producing adrenocortical carcinomas.
Further evaluation showed a left adrenal mass with enhancement features suggestive of a lipid-poor tumor. The characteristics of the tumor on adrenal CT scan will help distinguish a benign from a malignant lesion. Adrenal tumors with irregular margins more than 4 cm, with more than 10 HU and less than 50% washout point to a probable malignant lesion. An adrenal mass that measures less than 10 HU on unenhanced CT, has the density of fat, and is most likely a benign adenoma. However, up to 30% of benign adenomas are considered lipid-poor and may be indistinguishable from non-adenomas.[5] The patient’s adrenal mass had 26 HU and 65.4% washout, which were compatible with a lipid-poor tumor and therefore warranted further evaluation.
Complete hormonal work up is necessary in patients with suspected adrenocortical carcinoma even if clinical symptoms are absent not only for preoperative preparation but also because these can be used as markers for tumor recurrence. Autonomous hormone secretion can be expected in 80% of patients with adrenocortical carcinoma. There is a high likelihood that a large adrenal mass is not an adrenocortical cancer if autonomous steroid secretion is absent.[6] The pattern of hormone secretion may also point to the malignant potential of the lesion such as estradiol in males and virilization in females.[7] The patient had no feminizing symptoms such as decreased libido and erectile dysfunction but had mild gynecomastia on physical examination, which can be explained by his elevated estradiol levels. It is possible that the elevated estradiol level was due to aromatization of testosterone to estradiol in adipose tissue in an obese patient, however, normalization of estradiol after adrenalectomy confirmed that the tumor was secreting estradiol.
Although the patient had no signs and symptoms of hypercortisolism, hormonal evaluation showed elevated 24-hour urinary free cortisol, nonsuppressible cortisol after dexamethasone and decreased serum ACTH, consistent with subclinical Cushing’s syndrome. This can be associated with the risk of postoperative adrenal insufficiency. Due to the variable hypercortisolemia and the rapid development of adrenocortical carcinoma, clinical features of Cushing’s syndrome are often incomplete or even missing (atypical or subclinical Cushing’s syndrome).[8] Preoperative preparation therefore entails glucocorticoid coverage in patients with Cushing’s or subclinical Cushing’s syndrome. Preparation is also needed for patients with tumors secreting metanephrines, therefore it is important to exclude a pheochromocytoma prior to surgery. The patient’s 24-hour urine metanephrines were elevated but his plasma metanephrines were normal which made pheochromocytoma unlikely.
The histopathologic diagnosis of adrenocortical carcinoma is challenging as no single marker indicates malignancy. The Weiss criteria, with modification proposed by Aubert, is the most widely used diagnostic score. This includes high nuclear grade, >5 mitoses per 50 high-power fields, atypical mitotic figures, <25% of tumor cells are clear cells, diffuse architecture (>33% of tumor), necrosis, venous, sinusoidal and capsular invasion.[9] The presence of three or more criteria highly correlates with subsequent malignant behavior.
The patient’s tumor showed a high nuclear grade, frequent and atypical mitotic figures, diffuse architecture and areas suspicious for capsular invasion and therefore fulfilled the Modified Weiss criteria for malignant adrenocortical neoplasm (Figure 3). The provisional histopathologic diagnosis was malignant adrenal neoplasm (adrenocortical carcinoma versus pheochromocytoma). Immunohistochemistry stains were requested to differentiate between the two tumors. The immunohistochemistry stained positive for inhibin which confirmed the adrenocortical origin of the tumor. S100, a marker for neuronal origin, and chromogranin, a marker for neuroendocrine differentiation were negative, which ruled out pheochromocytoma. The Ki67 index which is a marker for proliferative activity can be used to determine the aggressiveness of the tumor. It is the most powerful prognostic marker in both localized and advanced adrenocortical carcinoma to guide treatment decisions6. A Ki67 index more than 10% indicates high-risk adrenocortical carcinoma and would require more aggressive treatment after surgery such as adjuvant therapy with mitotane.[8] The patient’s tumor had a Ki67 index of 5-10% which is classified as low-risk adrenocortical carcinoma.
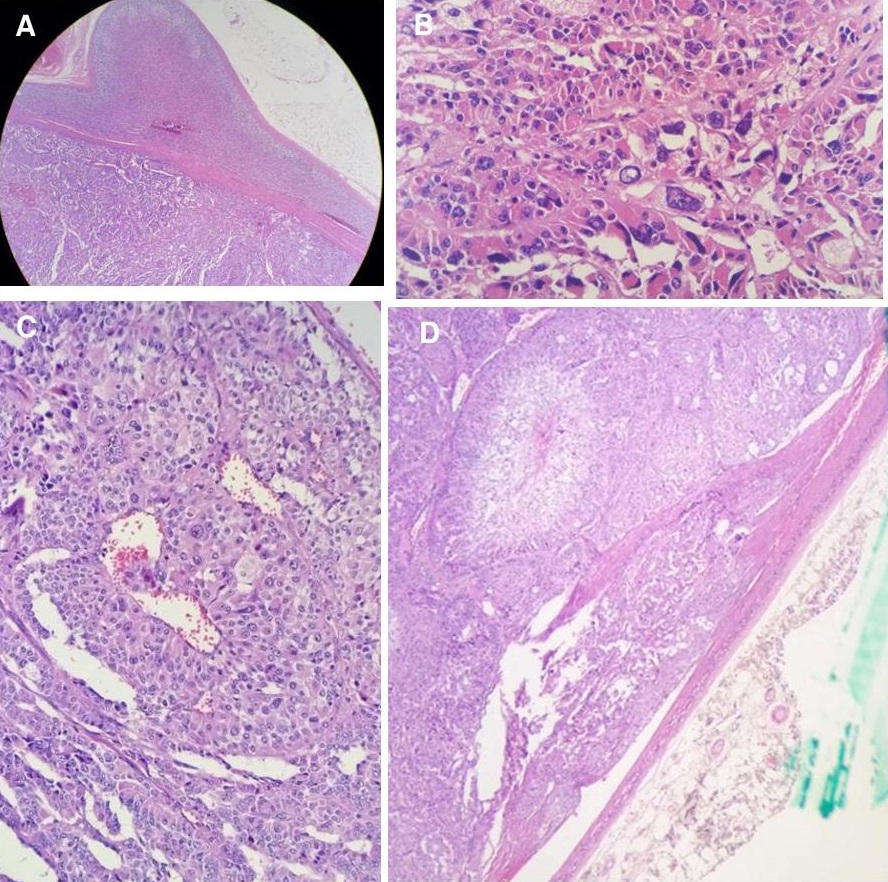
Figure 3. Histopathologic results of the left adrenal gland. (A) Uninvolved adrenal tissue and the encapsulated mass (H & E, 40x). The cells in the mass are arranged haphazardly compared to normal tissue. (B) Cells with marked pleomorphism, hyperchromatic nuclei and prominent nucleoli (H & E, 100x). (C) Areas with serpentine/ribbon-like arrangement of tumor cells with vascular channels within the tumor (H & E, 100x). (D) Areas suspicious for capsular invasion (H & E, 40x).
All patients suspected of having adrenocortical carcinoma should have a thoracic CT scan prior to surgery for proper staging.[8] The TNM classification is used in the assessment of disease stage. Stages I and II are described as localized tumors with a size of 5 cm or smaller and larger than 5 cm, respectively. Stage III includes locally invasive tumors whereas stage IV consists of tumors with distant metastases. The 5-year disease-specific survival rates are 82% for stage I, 61% for stage II, 50% for stage III, and 13% for stage IV disease.[6] Complete surgical resection of the tumor is the cornerstone of treatment in all patients having localized and locally-advanced disease. The resection status is a major predictor of prognosis and a margin-free complete resection provides the only means to achieve longterm survival.[6] Adrenalectomy was also reported to initially cure both hypokalemia and hypertension in most cases of aldosterone-producing adrenocortical carcinomas.[3] Open adrenalectomy is the standard surgical approach to patients with stages I to III adrenocortical carcinomas. Laparoscopic adrenalectomy may be done in patients with small tumors of less than 8 cm without preoperative evidence for invasiveness and suspected to be only potentially malignant.[8]
For patients with complete tumor resection, regular follow-up every 3 months with abdominal and thoracic CT scans and monitoring of initially elevated hormones are recommended. Interval follow-up may then be increased after 2 years and should be continued for at least 10 years thereafter.[7]
The patient underwent laparoscopic adrenalectomy with steroid coverage due to preoperative findings of hypercortisolism. He was classified as stage II based on the size of the tumor which measured 6 cm in its largest diameter. No further treatment such as adjuvant chemotherapy was done after complete surgical resection since his Ki67 of 5-10% was classified as low-risk. His blood pressure decreased, serum potassium, aldosterone, cortisol and estradiol levels returned to normal. However, he was still advised to follow up regularly for monitoring of tumor recurrence.
Aldosterone-producing adrenocortical carcinoma is a rare malignancy often presenting with uncontrolled hypertension and hypokalemia. In the work-up of suspected adrenal carcinoma, complete hormonal evaluation is necessary even if clinical symptoms are absent. Clinical features of hypercortisolism are often absent due to the variable hypercortisolemia and rapid development of adrenocortical carcinoma. It is important to exclude hypercortisolism and pheochromocytoma as these require preoperative preparation. The pattern of tumor secretion and tumor characteristics on CT scan may point to the malignant potential of the tumor. Complete surgical resection is the cornerstone of treatment. Ki67 index is the most powerful prognostic marker to guide treatment decisions. Long-term monitoring is recommended with imaging and hormonal evaluation used as tumor markers for recurrence.
Ethical ConsiderationPatient consent was obtained before submission of the manuscript.
Statement of AuthorshipAll authors certified fulfillment of ICMJE authorship criteria.
Author DisclosureAll the authors declared no conflict of interest
Funding SourceNone.
[1] United States. National Cancer Institute. Biometry Branch, Young JL, Cutler SJ, Connelly RR. Third national cancer survey: Incidence data. Bethesda, Md. : US Dept. of Health, Education, and Welfare, Public Health Service, National Institutes of Health, National Cancer Institute, 1975.