We described our experience in managing an uncommon case of autoimmune hypophysitis (AH) in a 58-year-old man. The initial clinical picture of quadruple anterior pituitary hormonal deficits and pituitary enlargement suggested the patient had macroadenoma but did not completely correlate with the presenting symptoms and the rate of evolvement to empty sella. The presence of AH in a man of this age group with a pituitary mass evolving to an empty sella within a short duration of seven months is rare. The etiology in this patient was a suspected autoimmune mechanism when we considered the unusual clinical course and the coincidental rate of regression of the enlarged pituitary to an almost empty sella.
CASEA 58-year-old man initially had a three-month history of persistent severe headache, lethargy and decline in sexual function with no visual and neurological deficit. There were no drenching night sweats, unexplained weight loss, fever, abdominal discomfort, swelling on the neck, axilla or groin, arthralgia and joint swelling, myalgia or myositis, shortness of breath or cough. His past medical history was unremarkable with no history of autoimmune disorders or endocrinopathy. The baseline early morning serum cortisol level was low (<22 nmol/L) (N: 68-469) with a normal serum adrenocortical hormone (ACTH) level of 5.0 pg/mL (N: 0-46.0). Despite the subnormal serum luteinizing hormone (LH) 0.9 IU/L (N: 2.0-12.0) and the follicle stimulating hormone (FSH) of 1.9 IU/L (N: 1.5-14.0) at the lower limit of normal, the corresponding serum testosterone level of 1.71 nmol/L (N: 9.9-52.4) was also low. In addition, the low level of serum TSH (0.86 uIU/L) (N: 032-5.0) response was inappropriate to the low serum free thyroxine (T4) level of 9.26 pmol/L (N: 9.10-23.80). There was also a low growth hormone (GH) level of <0.15 mIU/L (N: 0.16-16.0) with normal prolactin level of 2.04 ug/L (N: 1.61-18.77) at presentation. Formal perimetry showed no visual field deficit. There was no diabetes insipidus. Anti-thyroid peroxidase (TPO) antibodies level was not raised at 20.4 IU/mL (N: 0-35). The full blood picture did not show any histiocytes. Chest X-ray also showed no mediastinal enlargement, pulmonary infiltrates or hilar lymphadenopathy in both the lung fields. However, serum angiotensin-converting enzyme was not sent as there was no suggestion of sarcoidosis from our clinical assessment. Moreover, this assay was not available in our institution, and the patient could not afford the additional cost of sending to another laboratory with the facilities.
Initial magnetic resonance imaging (MRI) demonstrated a 20 mm by 14 mm by 10 mm non-homogenous mass occupying the pituitary fossa. This enlarged pituitary mass (white arrow in A) is associated with suprasellar extension (Figure 1). There is a T1 hyperintense rim around the wall of the mass, with some more amorphous hyperintensities at a dependent location within the lesion core. These T1 hyperintensities could represent either methemoglobin or proteinaceous content. Polypoidal T1 hypointensity within the sphenoid sinus anteriorly (white arrowhead in A), which enhanced post-gadolinium (not shown), is attributed to reactive inflammation. No appreciable enhancement of this mass was detected, nor was there evidence for a thickened pituitary infundibulum or hypothalamus. This initial MRI features of the enlarged pituitary, no thickening of pituitary infundibulum or hypothalamus, the negative clinical screening test for autoantibodies, sarcoidosis, tuberculosis, lymphoproliferative disorders and other granulomatous lesions suggested a macroadenoma.
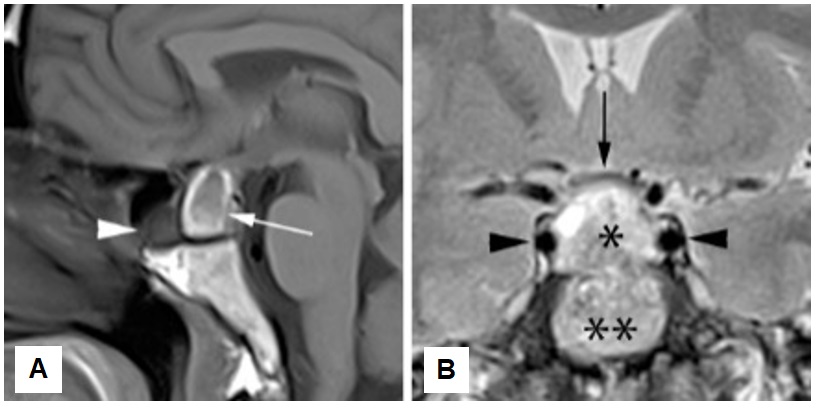
Figure 1. First pituitary MRI, T1-weighted midsagittal image without contrast (A) and T2-weighted coronal image (B). Heterogeneous pituitary mass but predominantly high signal at T2-weighted sequence (single asterisk in B). Its suprasellar component causes compression & mild upward displacement of the optic chiasm (black arrow in B). Double asterisks indicate the basisphenoid.
Correction of the hormonal deficit was the priority as our patient did not have compression of the optic nerve leading to visual field deficit. A physiological dose of hydrocortisone (10 mg am and 5 mg at noon) and oral levothyroxine of 75 mcg daily were commenced. Intramuscular testosterone 250 mg every three weeks was also initiated. His symptoms improved with the hormonal replacement therapy, which correlated with the improvement in his repeated hormonal profile. After the hormonal treatments for more than six months, the free T4 level improved to 16.72 pmol/L with only a minimal decline in the serum TSH level (0.27 uIU/L) and an improvement in serum testosterone of 49.8 nmol/L. His early morning serum cortisol level remained at <0.22 nmol/L. During this interval period, he denied any symptoms, such as syncope and severe headache, nor there was a new hormonal deficit to suggest pituitary apoplexy.
An MRI of the pituitary was repeated seven months following clinical improvement with hormone replacement therapy. Despite the absence of visual field deficit, reassessment of the size of pituitary mass is important for the determination of the subsequent treatment course. This follow-up MRI showed a drastic regression of the pituitary mass (Figure 2). While the T1 hyperintense “bright spot” of the posterior pituitary is appreciated (white arrowhead in A), much of the anterior pituitary appears to be flattened against the seller floor (white arrow in A). The optic chiasm prolapsed directly and inferiorly into the sella from its normal suprasellar location (black arrow in B), possibly reflecting traction due to post-inflammatory fibrotic process. Predominantly cerebrospinal fluid-filled pituitary fossa and the resolution of the inflammatory changes within the sphenoid sinus is also noted. A third MRI six months later revealed similar features of an empty sella. Formal perimetry assessments on both follow-up assessments did not show any visual field deficit.
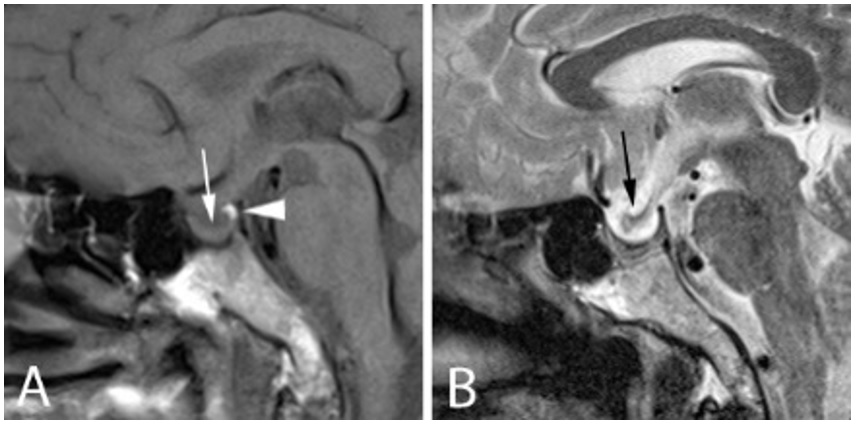
Figure 2. Follow up MRI in the midsaggital plane, T1-weighted (A) and T2-weighted images (B). The T1 hyperintense posterior pituitary (white arrowhead in A), with much of the anterior pituitary, flattened against the seller floor (white arrow in A). The optic chiasm is prolapsed inferiorly into the sella (black arrow in B).
The gender, the age of our patient at diagnosis and regressing pituitary mass to an empty sella without any neurological deficit or worsening of hormonal status is unusual.[1] We diagnosed AH clinically with the exclusion of other important causes of enlarged pituitary: lymphoma and granulomatous hypophysitis. Our patient was assessed and screened clinically with clinical history, physical examination and relevant biochemical, endocrinological, hematological and radiological investigations for the above problems. In addition, the clinical course of improved symptoms and well-being with a physiological dose of hydrocortisone as well as the initial inflammation with subsequent fibrosis in the empty sella (Figures 1 and 2), suggest an autoimmune inflammatory etiology in the pituitary. Moreover, the features of pituitary regression in the serial MRIs (Figures 1 and 2) appeared to be similar to the changes of AH induced in mice leading to pituitary atrophy and secondary empty sella.[2] In contrast, pituitary enlargement secondary to lymphoma or sarcoidosis have other clinical features and will require definitive chemotherapy or high dose corticosteroids respectively.
AH is a rare autoimmune inflammation of the pituitary gland. Majority, 90%, of AH are women (woman: man 6: 1-8: 1) with 90% premenopausal.[1],[3] There is a preponderance of women in late pregnancy and first six months of postpartum. The average age at diagnosis is 35 years for women and 45 for men.[1] Sporadic cases have also been reported in children, men, and the elderly.[4] No family or ethnic predisposition has been described. This rate of regression to empty sella in AH (three to seven months) has been reported in postpartum, young women and a younger man but not in a man of this age group.
The symptoms and hormonal deficits in our patient were consistent with adenohypophysitis, the most common manifestation of AH. In addition, the affected pattern of corticotrophs, gonadotrophs, somatotrophs, and thyrotrophs were also like most of the previously reported cases.[5] Other manifestations not found in our patient, for example, involving the infundibular stem, the posterior lobe of the pituitary gland (infundibuloneurohypophysitis) or the whole pituitary (panhypophysitis) are less common. In addition, adenohypophysitis preceded both infundibuloneurohypophysitis and panhypophysitis has also been reported.[3]
The presenting clinical features in our patient, his hormonal profile of the anterior pituitary and the secondary target organs suggested a primary pituitary dysfunction. Despite the low early morning serum cortisol level (<22 nmol/L), there was no paradoxical increase in the serum ACTH level which remained at 5.0 pg/mL. Similarly, the low level of serum free T4 (9.26 pmol/L), serum testosterone (1.71 nmol/L nmol/L) were not associated with a raised serum TSH level (0.86 uIU/L), serum LH (0.9 IU/L) and FSH (1.9 IU/L) respectively. This suggests secondary hypocortisolism (cortisol <22 mmol/L; ACTH 5.0 pg/mL), secondary hypogonadism (LH 0.9 IU/L, FSH 1.9 IU/L, serum testosterone 1.71 nmol/L), and secondary hypothyroidism (TSH 0.86 uIU/L, serum free T4 9.26 pmol/L) at baseline. A normal serum prolactin level (2.04 ug/L) ruled out a stalk effect of the pituitary gland. Taken together with the low serum GH level (0.15 mIU/L) this indicates an underlying primary dysfunction of the pituitary gland. Further, no improvement in the pituitary hormone responses over the follow-up periods suggested lack of recovery in the pituitary-primary organs hormonal axes.
The first MRI of this patient had findings suggestive of adenohypophysitis with no innfundibuloneurohypophysitis (Figure 1). Although MRI of the pituitary gland is the imaging technique of choice for the characterization of the lesions, identifying AH is still challenging.[6] In 75-90% of cases of AH, a pituitary mass was observed as in Figure 1, 70% showed ring-like enhancement after administration of contrast.7 In infundibuloneurohypophysitis, thickening of the pituitary stalk, with a diameter of more than 3.5 mm in the medial eminence of the hypothalamus is characteristic.[7] Marked enhancement of the stem after administration of gadolinium, which can extend to the lower end of the hypothalamus with a loss of hyperintensity of the neurohypophysis in the T1 sequence are also common characteristics. However, a hypointense posterior pituitary signal on T1 can also be observed in 10% of normal subjects.[8] In our patient, the serial hyperintense pituitary MRI images suggested a normal posterior pituitary and consistent with the absence of diabetes insipidus. Nevertheless, the absence of thickened pituitary stalk, the T1 hyperintense rim encased mass without contrast, and a radiological regression of the anterior pituitary to empty sella is unusual. In addition, even though the pituitary regressed to empty sella, there is no deterioration of the adenohypophysis hormonal pattern deficit in our patient.
Our patient did not present with any history of clinical autoimmune disorders, and the TSH and TPO antibodies were negative in this patient. Thus, the use of antibodies to facilitate the diagnosis of AH was unhelpful for this patient. TSH and TPO antibodies were sent because the presence of associated autoimmune diseases was reported in 20-50% AH, consisting mainly Hashimoto thyroiditis followed by Graves' disease.[9] Autoimmune adrenalitis was reported in 5-7% of cases, pernicious anemia and diabetes mellitus type 1 in 2%. Less frequently, association with autoimmune polyglandular syndrome type 2, vitiligo, mumps, lymphocytic disease, celiac disease, systemic lupus erythematosus and rheumatoid arthritis were also reported.
Antihypophyseal antibodies (against Α-enolase or specific neuronal enolase) was not sent as these assays were not available in the local laboratories. These antibodies may support the diagnosis in 24% of the cases[10],[11] but its sensitivity does not exceed 36%.[12] However, these antibodies are nonspecific and detected in other endocrinopathies such as Cushing's disease, pituitary adenomas, Sheehan’s syndrome, type 1 diabetes mellitus, Hashimoto's thyroiditis and Graves' disease. Therefore, antihypophyseal antibodies are only useful together with a thorough clinical assessment of the patient. As almost 80% of those patients with pituitary antibodies also have antibodies to thyroid gland or hormones,[12] thyroid antibodies may be surrogate markers in the absence of antihypophyseal antibodies assay. In our patient, thyroid antibodies screening was negative.
Although we did not have any histopathological specimen to confirm the diagnosis, our approach did not compromise the management of the patients. In the past, only two-thirds of patients with AH have biopsy confirmation.[3] Histopathological specimen from the biopsy specimen confirmed the nature of the lymphoplasmacytic infiltrate.[13],[14],[15] Biopsy also rules out other diseases that may have a similar clinical presentation. However, diseases such as tuberculosis, sarcoidosis and immunoglobulin deposits (IgG4) have other presentations which can be diagnosed with other safe investigative modalities.[11] In our patient, the absence of symptoms and signs along with the negative blood investigations and normal chest x-ray rule out the above conditions. Considering the potential risks of unnecessary invasive procedure of the pituitary, presumptive diagnosis combining context and clinical features can facilitate effective treatment. Confirmation of pituitary biopsy is not always necessary.[16] A case series suggested at the one, five and ten-year follow-up, there was no significant difference between the medically and surgically managed cohorts in term of ongoing symptomatology or need for pituitary hormone replacement.[17] Therefore, it is important to differentiate between AH and pituitary adenoma to avoid putting the patient at risk for unnecessary surgery (Table 1).
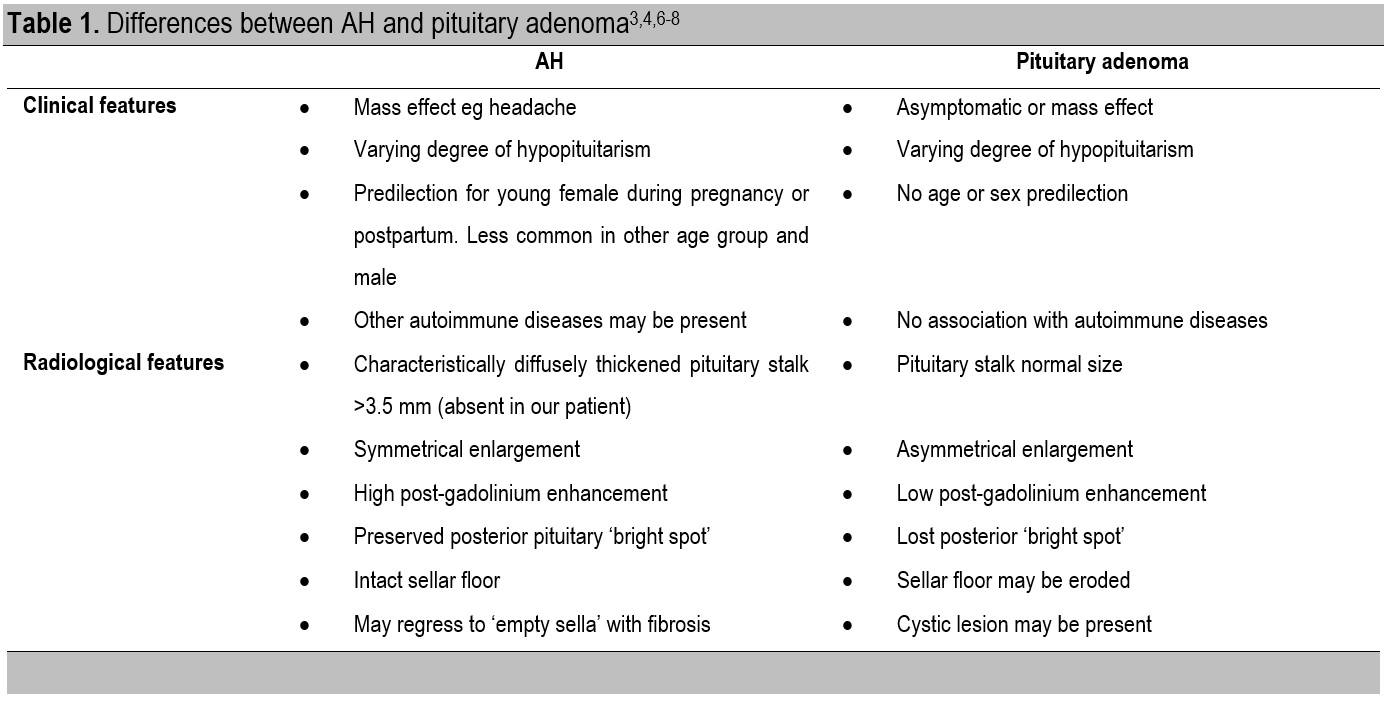
Table 1. Differences between AH and pituitary adenoma[3],[4],[6]-[8]
AH has a variable course, with no definitive treatment. Our patient receiving a physiological dose of hydrocortisone. High dose corticosteroids have been used to suppress the inflammation in AH,[3],[18].[19] and high doses of prednisolone were reported to be effective in reducing pituitary growth in 62.5% of the cases.[3],[20] Pulse methylprednisolone was most effective when the disease is less than six months.[21] Nevertheless, the number of AH is still too small for randomized controlled trials as well as characterization of the clinical features that respond to corticosteroid. Other authors have suggested immunosuppressive treatments such as methotrexate and azathioprine.[9],[18],[19],[21] Since our patient had drastic regression of the pituitary size, other immunosuppressive treatment was not indicated.
The prognosis of AH is variable, depending on the degree of inflammatory infiltration, duration, residual fibrosis and response to treatment.[1] In our patient, the clinical features, persistent yet stable pituitary hormonal deficit, was discordant with the regression of pituitary mass. Due to the uncertain clinical course, close, continuous active surveillance to monitor the long-term progression of the disease in our patient is required.
Although AH is extremely rare in a man in the late fifties, it must be considered in any sudden loss of anterior pituitary hormonal functions, and MRI findings of ring-enhanced varying hypointense mass. Since a biopsy for definitive diagnosis involved risks, a presumptive diagnosis of AH may be made based on clinical, laboratory, and radiologic information and a high index of suspicion. Exclusion of pituitary macroadenoma, lymphoma and granulomatous hypophysitis is also important. Prompt diagnosis will facilitate prompt treatment and rapid hormonal replacement by a multidisciplinary medical team to minimize the morbidity. Although there is documentation of variable rate of regression of pituitary mass to an empty sella in AH of younger premenopausal women, this rate of regression in a man of this age group has not been reported. Since the course of the disease for this patient is uncertain, close, continuous active surveillance along with prompt and appropriate management is essential.
Ethical ConsiderationPatient consent was obtained before submission of the manuscript.
Statement of AuthorshipAll authors certified fulfillment of ICMJE authorship criteria.
Author DisclosureAll the authors declared no conflict of interest.
Funding SourceNone.
[1] Bellastella G, Maiorino MI, Bizzarro A, et al. Revisitation of autoimmune hypophysitis: Knowledge and uncertainties on pathophysiological and clinical aspects. Pituitary. 2016;19(6):625-42. PubMed Crossref