Case Report of an Adult Female with Neglected Congenital Adrenal Hyperplasia (CAH)
Gayathri Devi Krishnan and Norhayati Yahaya
Gayathri Devi Krishnan, MD
Ministry of Health Malaysia
Hospital Raja Perempruan Zainab II
15586 Kota Bharu, Kelantan Malaysia
Tel. No.: +09-745-2000
Fax No.: +09-748-9651
E-mail: kgaya3@yahoo.com
ORCID: https://orcid.org/0000-0002-2384-3401
e-ISSN 2308-118x
Printed in the Philippines
Copyright © 2018 by the JAFES
Received July 24, 2018. Accepted August 24, 2018.
Published Online First: October 9, 2018.
An apparently well 27-year-old phenotypically male adult was seen at the endocrine clinic for gender assignment. Patient had been raised as a male and identifies as such. Abdominal CT scan showed a unilateral left adrenal mass and karyotyping revealed 46 XX female karyotype. She was diagnosed to have simple virilizing CAH and needed thorough counselling with subsequent management by a multidisciplinary team.
Keywords: Congenital Adrenal Hyperplasia (CAH), 17-hydroxyprogesterone, gender assignment
Congenital Adrenal Hyperplasia is an autosomal recessive disorder with 21-hydroxylase deficiency accounting for most cases.[1] Hydrocortisone is the treatment of choice in children but management of the adult patient remains controversial.[1] Few doctors are trained to manage adults with rare genetic conditions like CAH.[2] Adrenocortical tumors in CAH are not rare but are mostly benign.[3] This case report highlights the challenges in both physical and psychological management of an adult female with neglected CAH who has been raised as a male.
CASEA 27-year-old patient was referred to the adult endocrine team for gender assignment as the National Registration Department (NRD) required this information when she needed an identification card to apply for a driving licence. Patient was diagnosed with ambiguous genitalia at birth but was lost to follow-up. Antenatal period was uneventful and she remained relatively well thereafter, having no reason to seek medical attention. In primary school, she was the tallest in class, but shortest when in secondary school. Academic performance was average. She is the 6th of 7 siblings from a consanguineous marriage and has a brother who died suddenly at 2 months of age. She is 143 cm tall with mid-parental height of 154 cm. She exhibits external male body habitus and has a high pitched voice. Pubic hair was Tanner 4 with marked clitoromegaly measuring 3.5 cm, prominent labia majora and no palpable testes. She had never experienced vaginal bleeding and was not sexually active. Blood investigations revealed FSH-8.2 IU/L (1.79-22.5), LH-6.4 IU/L (2.12-12.86), testosterone-7.6 nmol/L (0.3-2.1), progesterone-32.1 nmol/L (<5-60), estradiol-448 pmol/L (84-1068), TSH-1.82 mIU/ml (0.4-4.0) and cortisol-138.2 nmol/L (193-772). Serum 17-OH progesterone level was >60.6 nmol/L (0.9-7.58) and cytogenetic analysis showed 46,XX apparently normal female karyotype. Abdominal CT scan showed a 4.1 x 4.7 cm left adrenal mass, 39-42 HU on plain scan that enhanced with contrast (Figure 1a and b). The right adrenal gland appeared normal. Bilateral ovaries and a small uterus were seen. After detailed assessment and discussion including consultation with the local religious authority, patient was assigned male gender given his upbringing and own preference.
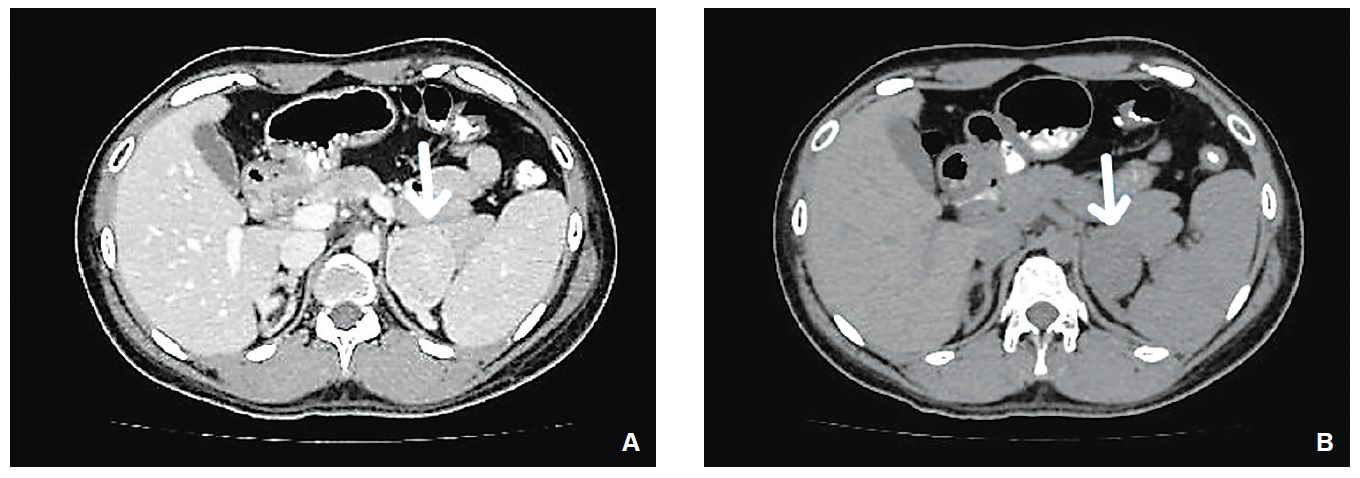
Figure 1a and b. Abdominal CT scan of the patient [(A) plain and (B) with contrast]. Arrows show a left adrenal mass.
Congenital adrenal hyperplasia (CAH) is an autosomal recessive disorder with 21-hydroxylase deficiency accounting for up to 95% of cases.[1] The clinical phenotype of CAH is typically classified into three, namely; classic salt losing, classic non salt losing (simple virilizing) and nonclassic forms. Although the same gene, CYP21A2, is involved, genetic mutations in the milder forms only partially impair 21- hydroxylase activity.[1] Treatment is aimed at replacing cortisol and aldosterone and controlling hyperandrogenism. Treatment of CAH especially in the adult patient, remains controversial.[1]
Our patient’s history suggests the simple virilizing form of CAH. She was lost to follow-up in early childhood leading to sub-optimal treatment, posing unique challenges in management as an adult. She has been well for the past 27 years with no hospital admissions; suggesting she never experienced adrenal crisis. A CT scan of the abdomen showed unilateral left adrenal hyperplasia with a left adrenal mass. She has been brought up as a male all her life and identifies as such given that she phenotypically exhibits a male body habitus. This raises the following issues; (i) the role of steroids, (ii) management of the adrenal mass and (iii) gender identification. We will address this in turn.
Androgen excess is always a problem for women with simple virilizing CAH, but the degree and clinical consequences vary.[2] At minimum, all affected women must receive physiological replacement doses of glucocorticoids.[2] Some patients, like ours, may do well for years without replacement dose glucocorticoids, but this predisposes them to life threatening adrenal crisis should they experience significant stress.[2] Higher doses of steroids are used to address hyperandrogenism if it is a concern.[2] Our patient was not troubled by the virilization and self identifies as a male. Physiological dose of steroids is recommended but treatment aimed at reducing hyperandrogenism may paradoxically cause more distress.
Chronic adrenal gland enlargement in CAH is associated with increased prevalence of adrenal tumors, including myelolipomas, especially if CAH is poorly controlled. It is believed to be due to chronic adrenocorticotropic hormone (ACTH) stimulation leading to adrenocortical cell metaplasia.[3] Reported prevalence rates of adrenocortical masses in homozygote patients are up to 83%. Adrenocortical masses may be bilateral or unilateral.[4] Majority are benign but adrenal carcinoma has been described previously in four cases with CAH.[4] Adrenal tumors in patients with CAH should be managed similarly to those without CAH. Our patient would need a detailed adrenal CT and possible surgical intervention should the mass be suspicious. However, despite every effort to convince her, she is not willing to undergo further tests or interventions as she feels well. This poses a challenge in the prognostication of the adrenal mass.
A major clinical manifestation of CAH is disorder of sex development (DSD), characterized by difficulty identifying gender from external genitalia as was the case with our patient. Gender identity is a sensitive issue carrying social, psychological and religious implications. Gender identity refers to an individual’s subjective internal sense of being a male or a female.[5] There are differing opinions about the time of gender assignment of a child with DSD; some psychologists suggest postponing it until after puberty and then to act according to the desire of the patient while other authors say it should be done according to the grade of virilization whereby a girl with more severe virilization should be raised as a boy.[6] A study by Maryam et al., in 2017 among patients with CAH suggested that karyotype did not have a role in gender identity. Hormonal disposition after birth had more effect on gender identity than intrauterine hormones and chromosomal parameters.[6] In cases of delayed presentation as with our patient, a meticulous discussion with the patient in the presence of a multi- disciplinary team should be undertaken. Gender assignment should ultimately be decided based on the patient’s perception and preference.[7] Continuous effort should be made in helping the patient through psycho-social and legal challenges that may be faced. Genital reconstruction surgery should be offered if appropriate. Our patient should have undergone thorough psychosocial assessment and counselling but this was no longer pursued due to her reluctance to attend further clinic visits.
Delayed diagnosis and management of CAH present a unique set of challenges that needs to be addressed based on individual assessment and approach.
Ethical ConsiderationThe authors submitted a letter of permission from the Head of the Department of Medicine of Hospital Raja Perempuan Zainab II to publish the case.
Statement of AuthorshipAll authors certified fulfillment of ICMJE authorship criteria.
Author DisclosureThe authors declared no conflict of interest.
Funding SourceNone.
[1] Merke DP. Approach to the adult with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J Clin Endocrinol Metab. 2008;93(3):653-60. PubMedPubMed Central CrossRef
[2] Auchus RJ. Management of the adult with congenital adrenal hyperplasia. Int J Pediatr Endocrinol. 2010; 2010:614107. PubMed PubMed Central CrossRef
[3] Teixeira SR, Elias PCL, Andrade MTS, Melo AF, Junior JE. The role of imaging in congenital adrenal hyperplasia. Arq Bras Endocrinol Metab. 2014;58(7):701-8. CrossRef
[4] Jaresch S, Kornely E, Kley HK, Schlaghecke R. Adrenal incidentaloma and patients with homozygous or heterozygous congenital adrenal hyperplasia. J Clin Endocrinol Metab. 1992;74(3):685-9. PubMed CrossRef
[5] Gender identity disorder in children. APA DSM-5 sexual and gender identity disorders. The International Foundation for Gender Education. https://www.ifge.org/302.6_Gender_Identity_Disorder_in_Children
[6] Razzaghy-Azar M, Karimi S, Shirazi E. Gender identity in patients with congenital adrenal hyperplasia. Int J Endocrinol Metab. 2017;15(3):e12537. PMID: 29201068. PMCID: PMC5701969. https://doi.org/0.5812/ijem.12537.
[7] Lee PA, Houk CP. Review of outcome information in 46,XX patients with congenital adrenal hyperplasia assigned/reared male: What does it say about gender assignment? Int J Pediatr Endocrinol. 2010;2010:982025. PubMed PubMed Central CrossRef
Authors are required to accomplish, sign and submit scanned copies of the JAFES Author Form consisting of: (1) Authorship Certification, that all the requirements for authorship have been met by each author, and that the final version of the manuscript has been read and approved by all authors; (2) the Author Declaration, that the article represents original material that is not being considered for publication or has not been published or accepted for publication elsewhere; (3) the Statement of Copyright Transfer [accepted manuscripts become the permanent property of the JAFES and are licensed with an Attribution-Share Alike-Non-Commercial Creative Commons License. Articles may be shared and adapted for non-commercial purposes as long as they are properly cited]; and the ICMJE form for Disclosure of Potential Conflicts of Interest. For original articles, authors are required to submit a scanned copy of the Ethics Review Approval of their research as well as registration in trial registries as appropriate. For manuscripts reporting data from studies involving animals, authors are required to submit a scanned copy of the Institutional Animal Care and Use Committee approval. For Case Reports or Series, and Images in Endocrinology, consent forms, are required for the publication of information about patients; otherwise, appropriate ethical clearance has been obtained from the institutional review board. Articles and any other material published in the JAFES represent the work of the author(s) and should not be construed to reflect the opinions of the Editors or the Publisher.